
A Unified Multiscale Theory of Animal Aging
A causal hierarchy that reorganizes aging biology into deep drivers, rate-control systems, amplifier loops, executor modules, and biomarker readouts.
Harry Negron¹
Biogerontology · Systems Biology · Published May 7 2026 · Full-text article
-
Harry Negron¹
¹ Jivaro Research, Nagaoka, Japan
Harry Negron is the author of this theory article and is responsible for the conceptual framework, literature synthesis, causal hierarchy, manuscript development, and figure/table planning. The article proposes a unified multiscale theory of animal aging centered on progressive loss of biological fidelity across molecular, cellular, tissue, systemic, and organismal layers.
Correspondence: h.negronpagan@gmail.com
-
© 2026 Harry Negron. Published by Jivaro Research. All rights reserved unless otherwise stated.
This article may be cited, linked, and quoted in brief excerpts for scholarly, educational, or review purposes with proper attribution. Reproduction, redistribution, or adaptation of substantial portions of the article requires permission from the author or publisher.
Jivaro Article ID: r7k4m2a
Key takeaways
Aging reflects progressive loss of biological fidelity across genome, epigenome, proteome, organelles, and multicellular signaling.bullet
IIS–mTOR–FOXO regulates aging rate, while senescence & inflammation convert damage into chronic pathology.
Clonal, microbial, mechanical, and endocrine-circulatory loops propagate local failure into tissue-specific decline, explaining reversibility and aging diversity.
Research Details
Topic: Biogerontology
Subtopic: Systems biology; geroscience; multiscale aging theory
Article type: Full-text theory article
Author: Harry Negron
Published: May 7, 2026
Updated: n/a
PDF: Available
Data appendix: No
Peer review status: Internal / External / Commentary / Review
Abstract
Aging research has identified recurring mechanisms but still lacks a ranked causal architecture. We argue that aging is best understood as progressive loss of biological fidelity across genome maintenance, epigenetic and chromatin organization, proteome quality control, organelle coordination, and multicellular signaling. Conserved nutrient- and stress-sensing pathways such as IIS–mTOR–FOXO regulate the pace of this loss rather than serving as its sole origin. As fidelity declines, protective responses including senescence and inflammatory signaling become chronic, while amplifier loops—including inflammaging, clonal hematopoiesis, dysbiosis, neuroendocrine-circulatory coupling, and tissue-mechanical drift—spread local failures into organism-wide state change. Tissue-specific executor modules then produce stem-cell exhaustion, cell-identity drift, and organ-specific decline. This ranked model explains more than flatter alternatives that treat hallmarks as co-equal lesions or equate clocks with mechanism. It also predicts that early shared changes will arise in chromatin state, proteostasis, and organelle communication, and that state-restoring interventions will improve clocks and function more readily than deep structural lesions.
Keywords
aging; biological fidelity; geroscience; systems biology; epigenetic information loss; proteostasis; inflammaging; senescence; stem-cell exhaustion; organ-specific aging; rejuvenation
Introduction: Why aging still lacks a causal theory
Aging biology has reached an unusual stage of maturity. The field no longer lacks candidate mechanisms; instead, it has an abundance of them. Over the past two decades, the dominant achievement of geroscience has been to identify recurrent processes that accompany, shape, or predict aging across organisms, tissues, and experimental systems. The hallmarks framework crystallized this progress by organizing aging into a shared set of mechanistic domains, and its recent expansion further refined the field’s map of recurrent aging biology [1,2]. Yet a map is not the same thing as a causal theory. The central problem is no longer whether genomic instability, epigenetic alteration, mitochondrial dysfunction, loss of proteostasis, cellular senescence, or altered intercellular communication matter. It is whether these processes can be placed into a causally ordered architecture that explains why aging emerges, how it propagates, and why it differs in tempo across tissues, species, and individuals [1,2].
This distinction matters because the current literature often places fundamentally different types of processes on the same conceptual plane. Some aging-associated phenomena are plausible deep causal drivers; others appear to be maintenance failures; still others are best interpreted as compensatory responses, self-reinforcing amplifier loops, tissue-level executors, or readouts. The hallmarks model remains indispensable precisely because it captures recurring biological features, but it does not by itself fully distinguish origin from response, or amplification from readout [1,2]. A mature aging theory must therefore do more than catalogue recurrent lesions. It must rank them.
The absence of such ranking has sustained a long-running ambiguity in the field. Aging is often described either as accumulated molecular damage or as a quasi-programmed biological process, yet neither description is sufficient on its own. Pure damage models struggle to explain why lifespan is so strongly regulable by conserved signaling pathways and why aging leaves highly structured molecular trajectories rather than only heterogeneous deterioration [3,5,18–20]. Pure program models, by contrast, risk overstating coordination and underestimating the role of stochastic insult, imperfect repair, and local tissue history [3,5]. The problem is not that one of these perspectives is wholly wrong. The problem is that both capture part of the truth while failing to specify the hierarchy that connects them. Aging may be coordinated without being centrally “programmed,” and it may be damage-driven without being reducible to passive wear.
Recent systems-level data sharpen this problem rather than resolve it. Single-cell and tissue atlases now show that aging is at once shared and heterogeneous: common age-associated patterns recur across organs, yet individual cell types and tissues diverge sharply in timing, magnitude, and mode of decline [96,97]. Human cell-aging atlases extend this logic further by showing that age-linked transcriptional remodeling is not random noise scattered uniformly across the organism, but a structured reorganization of cell states, lineages, and tissue environments [98]. Multi-omic analyses likewise suggest that aging may involve nonlinear transitions and coordinated state shifts rather than merely a linear accumulation of microscopic injury [100]. Taken together, these results argue that aging must be understood as a systems phenomenon—distributed across scales, but not therefore reducible to a bag of unrelated local failures [96–98,100].
At the same time, the emergence of biological-age measurements has made the conceptual gap in the field more obvious. DNA methylation clocks, pace-of-aging measures, and proteomic aging clocks can estimate biological age, mortality risk, disease burden, and organ-specific aging states with striking accuracy [18–20,109,110,114,115,120,121]. Their success demonstrates that aging leaves coherent, reproducible signatures in molecular state space. But that success also creates a temptation: to mistake a precise state estimator for a causal explanation. A clock may track aging exceptionally well while remaining agnostic about what generated the trajectory it measures. The current clocks literature is explicit on this point: predictive value does not, by itself, establish mechanistic primacy [4]. This epistemic distinction is essential. If the field confuses measurement with mechanism, it risks becoming increasingly accurate at describing aging while remaining incomplete about why aging occurs.
The existence of structured clocks nonetheless has theoretical importance. If aging were only the independent accumulation of uncorrelated microscopic insults, it would be harder to explain why age predictors generalize across tissues, and in some cases across mammals, as well as they do [18–20]. Conserved methylation structure across species implies that at least part of aging reflects constrained, repeatable changes in biological state rather than purely idiosyncratic failure [19,20]. Proteomic clocks and organ-aging signatures add to this picture by suggesting that whole-organism aging is expressed through partially coordinated organ-level states that predict disease and survival [114,115,120,121]. These findings do not prove a single upstream cause. But they do imply that any successful theory must explain how diverse local processes give rise to common, measurable organism-level trajectories.
The same logic applies to the growing literature on cell identity. Aging increasingly appears not only as damage, but as erosion of stable specification. In both atlas-scale and mechanistic studies, aged cells often display increased heterogeneity, inappropriate transcriptional programs, and weakened maintenance of lineage-relevant chromatin states [25,26,98]. This does not eliminate the importance of DNA lesions, mitochondrial defects, or proteotoxic stress. Rather, it suggests that the biologically visible expression of these insults may often be a loss of the organism’s ability to preserve accurate state across time. That idea has begun to converge with the “information loss” perspective in aging, which argues that aging may be better understood as degradation of the fidelity with which biological systems maintain genomic, epigenetic, proteomic, organellar, and multicellular organization [5]. If correct, this would provide a route for reconciling stochastic insult with structured aging trajectories: organisms age not simply because damage occurs, but because the systems that preserve reliable biological state become progressively less faithful.
What is missing, then, is not another list of aging mechanisms, but a framework that orders them. Such a framework must distinguish deep causal layers, primary maintenance layers, a rate-control layer, an antagonistic response layer, amplifier layers, executor layers, and a final readout layer for clocks and other state estimators [1,2,4]. Without that separation, the field risks conflating why aging starts, why it accelerates, why it becomes chronic, how it is amplified, and how it is measured.
This paper begins from the premise that aging remains theoretically incomplete because its dominant frameworks are richer in mechanism lists than in causal ordering. We therefore seek a unifying model that explains why aging is simultaneously conserved and variable, stochastic and structured, local and systemic, damaging and partially reversible. Our guiding hypothesis is that the field’s disparate observations can be integrated by treating aging as a progressive loss of biological fidelity across coupled molecular and multicellular layers, with conserved allocation pathways modulating the tempo of loss and self-amplifying systemic loops converting local failures into organism-wide decline. The purpose of the sections that follow is to test whether that architecture explains more of the evidence, across animals and with special relevance to humans, than the flatter alternatives now available [1–5,96–100].
What any successful aging theory must explain
Before proposing any unifying mechanism, it is necessary to define the empirical constraints that such a theory must satisfy. Aging biology now contains too many robust observations for a viable theory to explain only a favored subset. Any serious account of aging must simultaneously accommodate at least six facts: aging rate is regulable; aging tempo scales across species; aging is coordinated but tissue-specific; some aging phenotypes are reversible; exceptional longevity and negligible senescence exist in nature; and accelerated-aging states recapitulate recognizable portions of ordinary aging biology. A theory that fails on any one of these points may still describe part of aging, but it cannot claim to explain aging as a general biological process [2,4,5].
The first constraint is that aging rate is neither fixed nor purely passive. It is strongly regulable by both genes and environment. In one of the landmark discoveries in biogerontology, mutation of daf-2 doubled lifespan in C. elegans, establishing that longevity can be dramatically altered by perturbing a single conserved signaling pathway [40]. In mammals, rapamycin extended both median and maximal lifespan even when treatment began late in life, showing that aging rate remains pharmacologically modifiable well into adulthood [41]. At the primate level, caloric restriction improved health and survival in rhesus monkeys, reinforcing the conclusion that conserved nutrient-sensing and resource-allocation systems shape the pace of aging rather than merely its consequences [42]. Any theory that treats aging as immutable wear therefore starts from an empirically weakened position [40–42].
The second constraint is that aging tempo is not arbitrary across species. Comparative work shows that somatic mutation rate per year is strongly inversely related to lifespan across mammals, while the mutation burden at the end of life varies far less than lifespan itself [9]. In parallel, methylation-based age structure is conserved across mammals, and methylation rates at conserved age-associated sites scale negatively with maximum lifespan [19,20]. These observations matter because they imply that whatever aging is, it is not only local pathology unfolding independently within each species. It obeys species-level tempo constraints. A successful theory must therefore explain why both genetic damage accrual and epigenetic-state change slow down in longer-lived lineages, and why these two forms of temporal structure appear partially coupled rather than independent [9,19,20].
The third constraint is that aging is coordinated across the organism but not uniform across tissues. Single-cell atlases show broad age-associated changes across multiple organs while also revealing strong cell-type-specific and tissue-specific differences in timing and phenotype [96,98]. Regenerative decline studies likewise indicate that age impairs common processes such as stem-cell mobility and angiogenesis, but does so in tissue-dependent ways [99]. Multi-omic human profiling suggests that aging trajectories are nonlinear rather than smoothly linear, with distinct periods of molecular dysregulation emerging during adulthood [100]. Proteomic studies extend this logic to circulating state: organ-aging signatures can be estimated from plasma, accelerated aging in one organ predicts disease in that organ, and organ-specific clocks predict mortality and longevity across diverse populations [114,115,120,121]. GrimAge and DunedinPACE further show that whole-body biological aging is measurable as a coherent state variable associated with morbidity, disability, and mortality [109,110]. Any theory must therefore explain how aging can be at once systemic and heterogeneous: globally coordinated enough to yield clocks, yet locally diverse enough to produce different organ trajectories [96,98,100,109,110,114,115,120,121].
The fourth constraint is that at least part of aging is reversible. This is one of the most disruptive findings in the modern field because it sharply limits theories that treat aging as a one-way accumulation of irreparable wear. Partial reprogramming with OSK restored youthful DNA methylation patterns and transcriptomes in retinal ganglion cells, promoted axon regeneration, and reversed vision loss in aged mice and in a glaucoma model [22]. A later systemic AAV-based study reported that inducible OSK expression in very old wild-type mice extended median remaining lifespan and improved frailty-related measures [23]. Independent systemic-environment work showed that exposure of aged mice to young blood improved synaptic plasticity and cognition [104]. These findings do not imply that all forms of aging are reversible, but they do require any theory to distinguish reversible regulatory-state changes from irreversible structural lesions. A successful theory must therefore explain not only decline, but also selective rejuvenation [22,23,104].
The fifth constraint is that aging is not expressed identically across animals. Nature provides both slow-aging and apparently non-senescent systems. DNA methylation studies in bats show that longevity is negatively associated with the rate of age-related methylation change [79]. Naked mole-rats exhibit delayed aging phenotypes, resistance to age-related decline, and unusual maintenance of function across much of their lifespan [80]. Bowhead whales, among the longest-lived mammals known, show lineage-specific changes in genes linked to cancer, DNA repair, and aging [82]. Hydra is an even sharper challenge to simplistic universality: some Hydra species show constant mortality and fertility with age, and experimentally lowering autophagy in a normally non-aging Hydra is sufficient to induce an aging phenotype [84,85]. At the other end of the lifespan spectrum, the African turquoise killifish provides a naturally short-lived vertebrate model with a lifespan of only a few months [111]. These species do not invalidate a unified aging theory; they define the comparative boundary conditions it must satisfy [76,79,80,82,84,85,111].
The sixth constraint is that accelerated-aging states are informative precisely because they reproduce recognizable parts of normal aging biology. Progeroid syndromes such as Hutchinson–Gilford progeria syndrome and Werner syndrome load heavily onto nuclear architecture, DNA maintenance, and genome-stability pathways, indicating that these are not peripheral features of aging but deep vulnerabilities [86,88]. Cancer survivors frequently display frailty, senescence-related burden, and biological-age acceleration consistent with therapy-induced loading of DNA damage and senescence pathways [91]. Obesity is associated with increased risk of diseases tied to cellular-aging hallmarks and shares inflammatory and metabolic features with aging more broadly [93]. HIV-associated aging studies likewise point to accelerated epigenetic aging, telomere attrition, and chronic immune activation [95]. The critical inference is that these are not separate “fake aging” processes. They are perturbation models that overload particular layers of the same architecture that governs ordinary aging [86,88,91,93,95].
Taken together, these six constraints act as a filter for mechanism. Any successful aging theory must explain why aging is modifiable, why its tempo scales across species, why it forms coherent but tissue-divergent organismal states, why part of it is reversible, why some species suppress or delay it so effectively, and why accelerated-aging syndromes map onto recognizable parts of the same biology. This immediately rules out overly narrow accounts in which aging is only inflammation, only senescence, only telomere loss, only epigenetic drift, or only damage accumulation. Each of those views captures something real. None is sufficient unless it can be placed inside a broader architecture that satisfies all six constraints at once [2,4,5,24,25,77,78,83,90,92].
Why the hallmarks are necessary but not sufficient
The hallmarks framework remains the most successful conceptual synthesis in aging biology because it solved a real problem: it transformed a fragmented literature into a shared map of recurrent mechanisms. The original formulation identified a set of biological processes that recur across normal aging and age-related disease, and the expanded version updated that map to a broader 12-hallmark architecture that better reflects current evidence [1,2]. This achievement should not be understated. Without the hallmarks, attempts at unification would risk becoming speculative narratives detached from empirical regularities. The hallmarks are therefore necessary in the strongest scientific sense: any serious theory of aging must explain why these recurring processes appear so consistently across tissues, organisms, and interventions [1,2].
Yet the very success of the hallmarks framework has also exposed its limit. A map of recurrent features is not the same thing as a causal theory. The hallmarks identify what aging repeatedly involves, but they do not fully specify what is upstream, what is downstream, what is protective, what is propagative, and what is mostly a readout of state. This is not a flaw of the framework so much as a consequence of its purpose. The hallmarks were designed to organize the field, not to settle the question of causal primacy. As a result, the framework is empirically rich but ontologically mixed: it places fundamentally different kinds of biological entities on the same conceptual plane [1,2].
That mixing becomes obvious when one compares hallmark categories side by side. Some hallmarks are best understood as candidate deep causal layers, such as genome instability or epigenetic alteration [1,2]. Others are better interpreted as maintenance failures, such as loss of proteostasis or defective macroautophagy [2]. Still others behave more like antagonistic responses that begin as protective adaptations but later become pathological, as in cellular senescence [1,2]. Still others are primarily amplifier layers, especially chronic inflammation, dysbiosis, and altered intercellular communication, which can integrate many upstream perturbations into a common organism-wide syndrome [2,47–49,70,75]. And some aging-associated features are closest to integrative or executor states, such as stem-cell exhaustion and tissue-regenerative decline, where upstream processes become overt organ dysfunction [1,2]. When such heterogeneous processes are treated as a flat list, the resulting picture is biologically informative but causally under-resolved.
The hallmarks papers themselves already point in this direction. Both the original and expanded frameworks distinguish between more primary forms of damage, antagonistic responses, and more integrative forms of decline [1,2]. That partial hierarchy is an important strength and one reason the hallmarks remain foundational. But the categories remain broad, and they do not yet provide a sufficiently ranked architecture for a unifying theory. For example, “epigenetic alterations,” “cellular senescence,” “chronic inflammation,” and “stem-cell exhaustion” do not occupy equivalent causal positions. One may plausibly generate or intensify the next. Likewise, “altered intercellular communication” is less a singular origin than a convergence zone in which multiple upstream failures become tissue-extrinsic stress [1,2,47–49]. A theory that leaves these processes at the same conceptual depth risks confusing causal initiation with late-stage system behavior.
A second reason the hallmarks are insufficient is that they are not independent modules. They are densely coupled. Chronic inflammation can emerge from senescent cells, microbial dysbiosis, tissue damage, innate immune sensing, and declining immune regulation [47–49,70,75]. Stem-cell dysfunction can arise not only from intrinsic damage but also from inflammatory niche remodeling, metabolic imbalance, and systemic circulating-state changes [1,2]. Epigenetic alteration can reflect DNA damage responses, metabolic-state shifts, chromatin remodeling, or differentiation instability. Loss of proteostasis can worsen mitochondrial dysfunction, and mitochondrial dysfunction can in turn intensify proteotoxic stress and inflammatory signaling. In other words, the hallmarks are real, but they are not naturally separable compartments. They form a network. A flat framework can catalogue nodes in that network; it cannot, by itself, tell us which nodes are deepest sources, which are bridges, and which are terminal manifestations.
This problem becomes more acute in the era of systems data. Single-cell aging atlases show that aging is coordinated across tissues yet highly cell-type specific, implying that common upstream pressures are filtered through lineage-specific and tissue-specific architectures rather than simply expressed identically everywhere [96]. Multi-omic human studies further suggest that aging unfolds through structured trajectories and possible transition periods rather than as an even, linear decline [100]. These findings are difficult to reconcile with any view in which the hallmarks act as largely independent lesions accumulating in parallel. Instead, they imply that some hallmark-like processes are likely closer to system organization, while others are emergent expressions of broader state change [96,100]. The more strongly aging appears as a coordinated systems phenomenon, the less satisfactory a purely enumerative hallmark model becomes.
A third limitation is epistemic. The hallmarks are often used alongside biological-age clocks and multi-omic state estimators, but the relationship between the two is not conceptually clean. Clocks can integrate signals from many hallmarks at once and predict morbidity or mortality with striking power, yet prediction does not reveal which hallmark sits highest in the causal chain [4]. This matters because a framework that mixes mechanism categories without separating them from state estimators can inadvertently blur what aging is with how aging is measured. The clocks literature has made this distinction explicit: biomarkers of biological age are valuable precisely because they summarize system state, but they are not by default the system’s generative cause [4]. That warning extends to the hallmarks. Some hallmark-level features may be nearer to causal origin, while others may resemble emergent summaries of deeper instability.
There is also a theoretical limitation. The hallmarks framework is compatible with several competing broad interpretations of aging: accumulated damage, deregulated maintenance, maladaptive late-life program extension, loss of biological information, or combinations of these [1–5]. That plural compatibility is one reason the framework has been so broadly adopted. But for the same reason, the hallmarks do not decide among these interpretations. They describe what recurs; they do not yet explain why those recurrences belong to one underlying architecture rather than many partly independent ones. The information-loss perspective is useful here because it offers a way to reinterpret diverse hallmark categories as failures to preserve reliable biological state across multiple layers [5]. But even that perspective remains incomplete unless the hallmarks themselves are reordered into a structure that distinguishes deep fidelity loss from downstream amplification and execution.
For these reasons, the central methodological move of this paper is not to reject the hallmarks, but to reclassify them. We treat the hallmarks as the empirical starting set of recurring features, then sort them into causal layers: deep causal layers, primary maintenance layers, a rate-control layer, an antagonistic response layer, amplifier layers, executor layers, and a readout layer. In this reclassification, genome instability and epigenetic alteration become candidate deep causal layers; proteostasis and autophagy become primary maintenance layers; nutrient sensing becomes the rate-control layer; senescence defines the antagonistic response layer before later becoming causal in its own right; inflammation and dysbiosis occupy amplifier layers; stem-cell exhaustion and regenerative collapse occupy executor layers; and clocks remain measurement tools in the readout layer rather than root causes [1,2,4,47–49,70,75]. This does not eliminate ambiguity, but it turns a list of mechanisms into a framework that can be argued with, tested, and falsified.
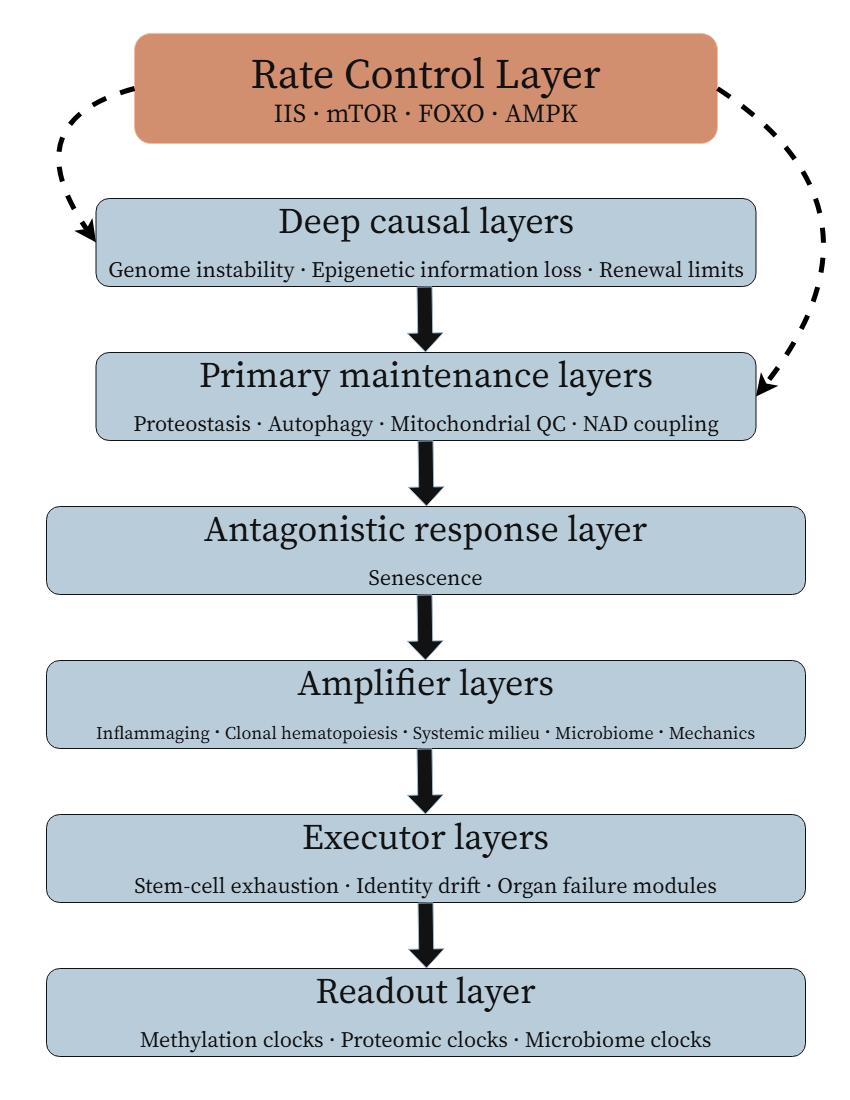
Figure 1. Ranked causal hierarchy of animal aging. Recurring aging mechanisms are reorganized here into causal roles rather than treated as a flat list. Deep causal layers generate progressive loss of biological fidelity; primary maintenance layers determine how well that fidelity is preserved in practice; conserved nutrient-sensing systems regulate the pace of loss; antagonistic responses such as senescence initially protect tissues but later become pathogenic; amplifier loops propagate local failures into organism-wide state change; executor layers determine organ-level phenotype; and clocks remain readouts rather than primary causes.
The payoff of this move is substantial. Once hallmark-like processes are treated as causal layers rather than flat categories, it becomes possible to ask sharper questions. Which processes should change earliest across many tissues? Which interventions should act broadly because they target higher-order layers? Why do some manipulations improve late-life function without reversing all forms of damage? Why do accelerated-aging states overload certain layers but not others? And why do long-lived species appear to preserve some aspects of the hierarchy more effectively than others? These questions cannot be answered well from a flat hallmark taxonomy. They require rank.
| Term | Meaning in this paper |
|---|---|
| Biological fidelity | Reliable preservation of information, identity, quality control, and coordination across scales. |
| Deep causal layer | Upstream sources of fidelity loss, especially genome, epigenome, chromatin, and renewal limits. |
| Primary maintenance layer | Systems that buffer fidelity loss, including proteostasis, autophagy, mitochondria, and NAD coupling. |
| Rate-control layer | Programs that set aging tempo, including IIS, mTOR, FOXO, AMPK, and dietary-response signaling. |
| Antagonistic response | Protective responses that become harmful when chronic, especially senescence and danger signaling. |
| Amplifier layer | Loops that spread local failure, including inflammation, clonal skew, dysbiosis, systemic milieu, and mechanics. |
| Executor layer | Tissue processes that produce overt dysfunction, including stem-cell exhaustion, identity drift, and organ failure. |
| Readout layer | Measurements of aging state, including clocks, organ-age scores, and mortality-risk predictors. |
Table 1. Core terminology of the proposed aging hierarchy. The table standardizes the major terms used throughout the paper and distinguishes causal layers from downstream readouts.
Thus, the hallmarks are indispensable but incomplete. They define the recurrent biological terrain of aging, but they do not yet supply the causal ordering required for a unified theory. Our task in the sections that follow is to preserve the empirical richness of the hallmarks while replacing their flatness with hierarchy. Only then can aging be understood not as a mere collection of associated lesions, but as a multiscale process in which some failures generate, some regulate, some amplify, some execute, and some merely report the organism’s progressive loss of biological fidelity [1,2,4,5,96,100].
Deep causal layer I: Genome instability, repair burden, and somatic evolutionary drift
Among the candidate deep causal layers of aging, genome instability occupies a special position because every other layer of biological organization depends, directly or indirectly, on the faithful preservation and regulated use of DNA. This does not mean that aging can be reduced to mutation accumulation alone. The stronger and more defensible claim is that genomic lesions impose a class of constraints unlike most other molecular insults: they alter sequence, disrupt replication, trigger checkpoint programs, recruit chromatin-remodeling machinery, reshape transcriptional activity, and create heritable differences among neighboring cells. For that reason, genome instability is best viewed not as one hallmark among many interchangeable hallmarks, but as one of the deepest pressures that long-lived animals must continually resist if they are to preserve coherent biological state across time [6,7].
The descriptive case for this is now strong. Normal human tissues accumulate somatic mutations with age, and they do so in tissue-specific ways that reflect differences in replication history, local environment, metabolism, and DNA-repair context [8]. Aging tissues are therefore not merely older versions of their youthful selves; they become progressively more mosaic. What begins as a genetically near-uniform soma increasingly resolves into a patchwork of related but non-identical cellular genomes. This observation matters because it changes the ontology of aging. Aging is not simply a uniform decline imposed on genetically identical cells. It is also the emergence of intra-organismal genomic heterogeneity, which creates new possibilities for altered lineage behavior, altered stress responses, and altered tissue ecology [8,10].
Comparative biology makes this constraint even harder to ignore. Across mammals, somatic mutation rate per year scales inversely with lifespan, such that long-lived species accumulate mutations more slowly than short-lived ones [9]. This result is one of the most important comparative findings in modern aging biology because it places genome maintenance directly into the lifespan problem. A successful theory must explain why evolutionary increases in lifespan are accompanied by slower genomic destabilization per unit time. Just as important, the same study implies that mutation burden at the end of life varies much less than lifespan itself, which cautions against a simplistic reading in which aging is merely the linear counting-up of sequence changes [9]. The inference is subtler and more consequential: lifespan appears constrained not just by how much genomic damage can be tolerated, but by how quickly genomic insult, repair, and downstream selection accumulate across the life course.
That distinction is crucial. If mutation count alone were the decisive variable, then similar terminal burdens across species should imply similar aging phenotypes, which they do not. The more plausible interpretation is that the biologically relevant variable is composite: lesion load, pathway-specific repair fidelity, chromosomal and structural instability, mutational spectrum, and the cumulative burden imposed by repeatedly detecting and resolving DNA damage [7,9]. In other words, DNA damage matters not only when it leaves a permanent lesion behind, but also because each episode of genomic insult mobilizes an expensive and potentially state-disruptive response. Double-strand break sensing, checkpoint activation, fork stabilization, transcriptional pausing, chromatin opening and reassembly, and selective deployment of repair pathways all impose energetic and regulatory costs on the cell [7]. Young organisms can absorb that cost. Aging organisms may increasingly pay for it in compromised state maintenance.
This point leads to a broader theoretical move. Genome instability should not be interpreted solely as an inventory of unrepaired lesions. It should also be interpreted as a continuing source of repair burden. Even “successful” repair is not a neutral event. The cell must transiently suspend normal priorities, alter chromatin accessibility, recruit repair complexes, and sometimes reconfigure transcriptional or replicative programs in order to preserve genomic function. Over decades, especially in stem cells and long-lived differentiated cells, repeated invocation of these programs may itself become a route by which biological information is eroded [5,7]. This inference aligns with later evidence that genomic disturbance can spill into innate immune activation through cytosolic DNA sensing and transposable-element derepression, suggesting that DNA instability burdens not only the genome but the regulatory and inflammatory architecture of the cell [50,54,55]. Thus, even before one invokes later amplifier loops, DNA damage is already more than damage.
Once aging tissues become genomically mosaic, a second process enters: somatic evolution. Clonal expansion in normal tissues shows that aging is not just the passive storage of mutations in isolated cells, but the selective amplification of some damaged lineages over others [10]. The importance of this point is often underestimated because somatic evolution is still too often viewed through the lens of cancer alone. Yet the broader aging relevance begins earlier and extends further. A clone need not become malignant to matter. It may simply proliferate more effectively, resist apoptosis, better tolerate stress, occupy more niche space, or produce altered secretory signals. Over time, such clones can remodel tissue composition, displace competing lineages, and alter local function without ever crossing the formal boundary into neoplasia [10]. Aging tissues are therefore not only damaged tissues; they are also evolving tissues.
The clearest human example of this logic is clonal hematopoiesis. Age-related clonal hematopoiesis is common in otherwise healthy individuals and predicts hematologic malignancy, cardiovascular disease, and all-cause mortality [11]. This immediately elevates somatic evolution from a cancer-specific concern to a central component of aging biology. More importantly, the evidence is now causal as well as associative. TET2-deficient hematopoietic clones accelerate atherosclerosis in mice by shifting macrophage inflammatory behavior in a pathogenic direction [12]. The loop is bidirectional: inflammatory environments, including TNFα-rich conditions, can favor the expansion of Tet2-mutant clones [13]. Here the significance of genomic mosaicism becomes unmistakable. Mutation does not merely accumulate; it changes the composition of the tissue, and the altered tissue state feeds back to select the very clones that intensify dysfunction. This is aging as somatic ecological drift.
These observations also clarify why genomic instability belongs near the base of the hierarchy rather than being treated as a late accompaniment of decline. When DNA damage and clonal selection intensify, the consequence is not simply more molecular disorder. It is a change in the rules by which cells persist, compete, differentiate, and communicate. In that sense, genomic instability is generative: it creates new cell-intrinsic states and new tissue-level selection landscapes. The organism becomes less a coordinated population of cells with a shared genomic operating frame and more a consortium of increasingly divergent lineages, each shaped by its own mutational history and competitive context [8–13]. The deeper significance of this divergence is that it threatens multicellular coherence itself.
Premature-aging syndromes reinforce this conclusion. Hutchinson–Gilford progeria syndrome, driven by LMNA/progerin-associated defects in nuclear architecture, produces chromatin abnormalities, genomic stress, defective DNA damage responses, and a strikingly compressed multi-system aging-like phenotype [86,87]. Werner syndrome, caused by deficiency of the WRN helicase, imposes replication stress, defective DNA maintenance, genomic instability, and telomere dysfunction, again yielding a broad premature-aging syndrome [88,89]. These conditions are not perfect replicas of normal aging, and they should not be treated as if they were [90]. But they remain highly informative perturbations. When the genome-maintenance and nuclear-architecture layer is overloaded, many aging-like features emerge rapidly and systemically. That is precisely what one would expect if this layer were close to the foundations of organismal aging.
The lesson of progeroid states is therefore not that ordinary aging is simply a mild version of one rare genetic disease. It is that normal aging and premature-aging syndromes overlap in vulnerable architecture. Both reveal that multicellular life depends on maintaining genomic integrity, repair competence, and nuclear structural coherence over time. Where progeroid syndromes differ is in how selectively and aggressively they load these vulnerabilities [86–90]. Their incompleteness is not a weakness for the present argument; it is a strength. It shows that overloading one deep causal layer can reproduce much of aging without reproducing all of it, exactly as one would expect in a layered causal hierarchy.
Taken together, the evidence supports a stronger interpretation than the familiar phrase “DNA damage accumulates with age.” Genome instability is a deep causal layer because it does three things at once: it seeds irreversible sequence and structural changes, it imposes repeated repair-associated burden on regulatory systems, and it creates substrates for somatic selection within tissues [6–13]. In this view, DNA damage is not merely passive debris stored inside old cells. It is a continuing generator of divergence within the organism and a persistent source of pressure on the mechanisms that maintain coherent cell state. This is why genome instability belongs near the base of the aging hierarchy—not as a solitary master cause, but as one of the deepest sources from which later epigenetic distortion, inflammatory propagation, and regenerative failure can emerge [5,6,7,50,54,55]. The next question, and perhaps the more unifying one, is what repeated genomic insult does to biological information itself.
Deep causal layer II: Epigenetic information loss and erosion of cell-state fidelity
If genome instability is one of the deepest sources of aging pressure, then epigenetic information loss may be the more general language in which that pressure becomes biologically visible. DNA sequence alone is not enough to explain multicellular life. Cells with the same genome become neurons, hepatocytes, lymphocytes, and myofibers because they maintain different chromatin configurations, transcriptional programs, and lineage memories over long periods of time. Aging therefore cannot be understood only as damage to the genome’s letters; it must also be understood as progressive failure to preserve the correct interpretation of those letters. In this sense, the epigenome is not a decorative overlay on the genome. It is part of the machinery that keeps cell identity stable, gene expression context-appropriate, and tissues functionally coordinated across time [5,25].
The strongest empirical clue that this layer is central is the extraordinary structure of age-associated methylation change. DNA methylation patterns track chronological age across a wide range of human tissues and cell types, indicating that aging leaves reproducible, non-random signatures in epigenetic state [18]. That structure is not confined to humans. A universal mammalian methylation framework has shown that age-linked methylation patterns are conserved across species [19], and methylation rates at conserved age-associated CpGs scale negatively with maximum lifespan across mammals [20]. These findings are difficult to reconcile with a view in which aging is only the uncoordinated accumulation of independent local lesions. Instead, they imply that aging involves constrained trajectories through epigenetic state space. That conclusion is reinforced by second-generation methylation measures such as GrimAge and DunedinPACE, which predict healthspan, mortality, and pace of aging rather than merely chronological age [109,110]. As emphasized earlier, clocks are not identical to mechanism [4]. But their structure and predictive power strongly suggest that aging includes a conserved, biologically meaningful reorganization of regulatory state [18–20,109,110].
What, then, is “epigenetic information” in the aging context? It is not best conceived as a static list of methylation marks or histone modifications. It is better understood as the cell’s capacity to repeatedly reconstruct the appropriate chromatin architecture, transcription-factor access landscape, and lineage-specific regulatory logic after division, stress, repair, and environmental fluctuation [5,24,25]. In youth, this restoration is sufficiently faithful that tissues preserve identity and function despite constant perturbation. In aging, that restoration may become noisier, less precise, and more path-dependent. The consequence would not necessarily be immediate cell death. It would instead be progressive erosion of fidelity: cells remain alive, but increasingly fail to maintain the exact regulatory state that defines what they are supposed to be and how they are supposed to respond.
This interpretation gains real mechanistic force from the evidence that chromatin disruption itself can drive aging-like phenotypes. In a key study, inducible double-strand breaks that did not primarily act by increasing mutational burden were nonetheless sufficient to accelerate aging-like changes by eroding epigenetic information [21]. The importance of this result is hard to overstate. It directly links the repair burden discussed in the previous section to a deeper regulatory consequence: even when the genome is not catastrophically rewritten at the sequence level, repeated DNA damage responses may still destabilize the chromatin framework through which the genome is read [21]. In this view, damage and information loss are not competing explanations. The former may be one route into the latter. What ages may not simply be the genome as a string of bases, but the cell’s ability to restore the correct chromatin state after having to defend that genome over and over again.
Independent work supports the idea that aging can impose altered chromatin states that are not irreversibly fixed. A recent study reported that aging establishes a hyper-quiescent chromatin state that can be reversed by regeneration [116]. This matters because it suggests that age-linked chromatin change is not merely degenerative residue; it can behave like a stabilized but plastic state. That observation immediately makes aging more compatible with partial reversibility and less compatible with a purely one-way wear model. It also provides an important conceptual bridge: if genome instability threatens fidelity from below, chromatin state may be the medium in which fidelity is lost, stored, and in some cases restored.
The rejuvenation literature is especially informative here. Partial reprogramming with OSK restored youthful epigenetic information and improved function in retinal ganglion cells without wholesale cell replacement, while a later systemic study reported lifespan extension and reversal of age-related changes in very old mice [22,23]. These findings do not prove that epigenetic change is the sole or first cause of aging, and the recent reprogramming literature correctly cautions that rejuvenation must be distinguished from uncontrolled dedifferentiation [24]. But they do show something essential: at least part of what ages is encoded in a regulatory layer that remains recoverable within old cells themselves. If an aged cell can regain youthful function after partial resetting of regulatory state, then aging cannot be understood only as a pile of irreversible lesions. Some central component of the phenotype must lie in how biological information is organized and retrieved [22–24]. That fact is one of the strongest arguments for placing epigenetic information loss near the base of the hierarchy.
The cellular phenotype of aging is increasingly consistent with this view. Large-scale atlas work suggests that aging includes loss of immune-cell identity, increased transcriptional heterogeneity, and weakening of the molecular boundaries that normally separate stable cell types [26]. A recent human cell aging transcriptome atlas extends that picture by showing widespread age-associated remodeling of transcriptional states across human tissues [98]. In parallel, mechanistic studies point to concrete routes by which identity can erode. In muscle stem cells, depletion of S-adenosylmethionine drives loss of heterochromatin and functional aging, and metabolic restoration can partially rejuvenate these cells [27]. In the aging B-cell lineage, higher-order chromatin reorganization contributes to defective development and altered lineage control [28]. Taken together, these studies suggest that aging is often not simply a transition from health to damage, but from stable specification to increasingly unstable specification. Cells do not only become impaired. They become less reliably themselves.
This shift in perspective also helps reinterpret “plasticity” in aging. The relevant question is not whether aging cells become generically more plastic or less plastic in a simple sense. Rather, aging appears to distort the balance between stable identity and appropriate adaptability [25]. Some cells may become locked into maladaptive states; others may drift into inappropriate stress programs or lineage-incongruent expression patterns. In either case, the common denominator is loss of fidelity in state maintenance. This is why cell-state erosion may offer a more unifying language than many classical hallmark categories. Mitochondrial stress, proteostatic burden, inflammatory signaling, and DNA damage can look different at the point of origin, yet they may converge on a shared outcome: weakening of the regulatory architecture that tells a cell what it is and how it should behave in context.
Recent evidence on mesenchymal drift makes this possibility especially provocative. A 2025 study reported prevalent mesenchymal drift across aging and disease states and showed that this drift could be reversed by partial reprogramming [119]. If this generalizes, it suggests that one common macroscopic expression of epigenetic and identity erosion may be movement toward a mesenchymal-like stress-adaptive state. Such a state could be advantageous in the short term, as a survival or repair posture, yet maladaptive when chronically stabilized. This interpretation remains more speculative than the methylation and reprogramming evidence, but it is theoretically important because it offers a candidate shared phenotype of regulatory decline across organs [119]. Instead of every tissue aging by unrelated programs, diverse tissues might converge on partially overlapping state drifts shaped by damage, inflammation, mechanics, and local niche context.
The attraction of this layer as a unifying candidate is therefore not that it replaces all other mechanisms, but that it can explain more of their convergence than they can explain of one another. Epigenetic information loss can accommodate the structured nature of aging clocks [18–20], the cross-species scaling of aging tempo [19,20], the reversibility seen in partial reprogramming [22–24], the regenerative reversibility of aged chromatin states [116], and the growing evidence that aging cells are often mis-specified rather than simply broken [26–28,98,119]. It also fits naturally with the previous section: genome instability and repair burden may be among the major upstream forces that erode this information. In that sense, the deepest unifying candidate is not damage alone, but damage-driven loss of biological information.
That said, caution is still required. Not every epigenetic change observed with age is necessarily causal. Some may be compensatory, some tissue-specific, and some downstream summaries of other insults [4,24]. The argument here is therefore comparative rather than absolute. Among the candidate deep causal layers of aging, epigenetic information loss currently explains more of the field’s hardest observations—structured state trajectories, partial rejuvenation, tissue-specific divergence, and identity erosion—than any purely lesion-based account can explain by itself. For that reason, chromatin-state instability deserves to be treated not as a downstream curiosity, but as one of the strongest candidates for the deepest common layer of animal aging. The next question is how this generalized loss of fidelity interacts with a more specific and lineage-dependent constraint: the proliferative limits imposed by telomeres and renewal tempo.
Deep causal layer III: Renewal tempo, telomere attrition, and proliferative system limits
If genome instability and epigenetic information loss define two of the deepest pressures in aging, telomere attrition introduces a third kind of constraint: a structural limit tied to cellular renewal itself. Telomeres are unusual among aging mechanisms because they translate division history into a physical boundary condition. They therefore do not act like a general background stress present equally in all cells at all times. Instead, they matter most where biological function depends on sustained replication—stem-cell systems, progenitor compartments, immune renewal, and high-turnover tissues. For this reason, telomere attrition belongs near the base of the hierarchy, but in a more conditional way than genome instability or chromatin-state erosion. It is a deep constraint on some of the most important tissues in vertebrate aging, yet it is not obviously the universal denominator of animal senescence [2,14,15].
The classical experiments remain foundational. In serially passaged human fibroblasts, telomeres shorten with progressive replicative aging, providing a direct molecular correlate of the Hayflick limit [14]. Follow-up work showed that telomere length predicts remaining replicative capacity, strengthening the case that telomeres are not merely passive markers of time in culture, but part of the mechanism by which proliferative potential is bounded [15]. These findings established one of the clearest routes by which aging can emerge from repeated normal function: the very act of renewing tissue exacts a structural cost on chromosome ends. In proliferative systems, aging is therefore not only a problem of accumulated insult or regulatory drift; it is also a problem of how many times a lineage can renew while preserving functional competence.
This places renewal tempo at the center of the issue. Tissues that depend heavily on ongoing cell replacement ask more from their stem and progenitor pools than tissues that are largely post-mitotic. The stem-cell literature has long emphasized that aging and regeneration are inseparable, because tissue maintenance is not achieved once during development but must be re-achieved continuously throughout life [58]. In that context, telomeres can be viewed as one major bookkeeping device of renewal burden. The faster a compartment turns over, the more strongly its long-term integrity depends on how stem cells ration proliferation, preserve quiescence, and maintain replicative reserve. Telomeres are thus especially informative because they make explicit that aging is not only about molecular fidelity within cells, but also about the long-term economics of replacing cells at all.
At the same time, the biology of renewing systems shows why telomere attrition cannot be treated as a complete explanation on its own. Exposure of aged progenitor cells to a young systemic environment can restore regenerative capacity, demonstrating that stem-cell aging is not reducible to an irreversible cell-intrinsic counting process [57]. More recent work in hematopoiesis further complicates any purely telomere-centric view. A mitochondrial unfolded protein response checkpoint regulates hematopoietic stem-cell aging [61], and depletion of myeloid-biased hematopoietic stem cells can rejuvenate the aged immune system [62]. These findings imply that proliferative aging is shaped not only by replicative history, but also by quality-control programs, lineage bias, systemic milieu, and niche context. Telomeres matter in these systems because they constrain renewal, but the actual age phenotype emerges from their interaction with broader fidelity-maintenance and state-regulation layers.
The strongest causal support for the importance of telomeres comes from reversibility experiments. In aged telomerase-deficient mice, telomerase reactivation reversed degenerative phenotypes in multiple tissues, implying that at least a substantial subset of telomere-driven aging features remains recoverable when telomere maintenance is restored [16]. Telomerase gene therapy in adult and old mice likewise delayed aging and extended longevity without an obvious increase in cancer in that setting [17]. These are powerful results because they show that telomere dysfunction is not merely correlated with age but can participate causally in organismal decline. They also reinforce a broader point made throughout this paper: some age phenotypes are not terminal endpoints of wear, but stabilized states that can move in a more youthful direction when a critical maintenance layer is restored.
Yet these same results also teach restraint. Telomerase-deficient mice are useful precisely because they exaggerate telomere pathology; they reveal what happens when one specific deep causal layer is overloaded, not necessarily what explains all normal aging [16]. Even the gene-therapy findings in otherwise aging mice, important as they are, do not justify making telomeres the singular master cause of senescence [17]. Many aging phenotypes arise prominently in post-mitotic tissues or in systems where altered signaling, proteostatic stress, mitochondrial dysfunction, immune activation, or epigenetic drift are already central. Even within the immune system, where renewal is crucial, early involution of organs such as the thymus reflects architectural and developmental decline that cannot be reduced to telomere length alone [52]. The lesson is not that telomeres are peripheral; it is that they are decisive in particular compartments and partially explanatory across the organism, rather than universally sufficient.
Comparative biology strengthens this qualified placement. The existence of animals with negligible senescence, such as Hydra, argues against the idea that all aging must be driven by a universal replicative clock. In Hydra, aging-like decline can be induced through failure of autophagy and epithelial stem-cell maintenance, showing that long-term tissue persistence can be destabilized through mechanisms other than classical telomere-driven replicative exhaustion [84,85]. This does not mean telomeres are irrelevant outside mammals, only that the general animal-aging problem cannot be collapsed into telomere attrition without remainder. The broader comparative lesson is that proliferative limits matter most where life history and tissue design depend on finite, tightly regulated renewal pools; they matter less as a stand-alone explanation for all forms of animal aging.
Telomere biology therefore fits best as a lineage- and tissue-specific deep causal layer within a broader fidelity-loss model. It captures something fundamental that the previous sections do not: renewal itself carries age-related liability. A tissue can lose function not only because its cells accumulate damage or drift from proper epigenetic state, but because the act of replacing cells gradually consumes structural reserve. In renewing compartments, telomeres turn time and replication into a measurable boundary. But the fact that progenitor function can be rejuvenated by systemic context [57], that stem-cell aging depends on mitochondrial and lineage-control checkpoints [61,62], and that some animals suppress aging-like decline through other maintenance systems [84,85], all argue that telomere attrition is one boundary condition among several, not the universal source of aging.
In the framework developed here, telomeres occupy an important but carefully delimited place. They are not a rival to genome instability or epigenetic information loss so much as a special case of how fidelity can fail in renewing systems. Telomere shortening constrains the longevity of lineages; telomere dysfunction can drive degeneration; telomerase restoration can reverse part of the phenotype. But telomere biology becomes most intelligible when placed inside the larger architecture of aging: genome damage and repair burden threaten fidelity from below, epigenetic drift destabilizes identity, and telomeres impose a renewal limit on the very compartments responsible for tissue maintenance.
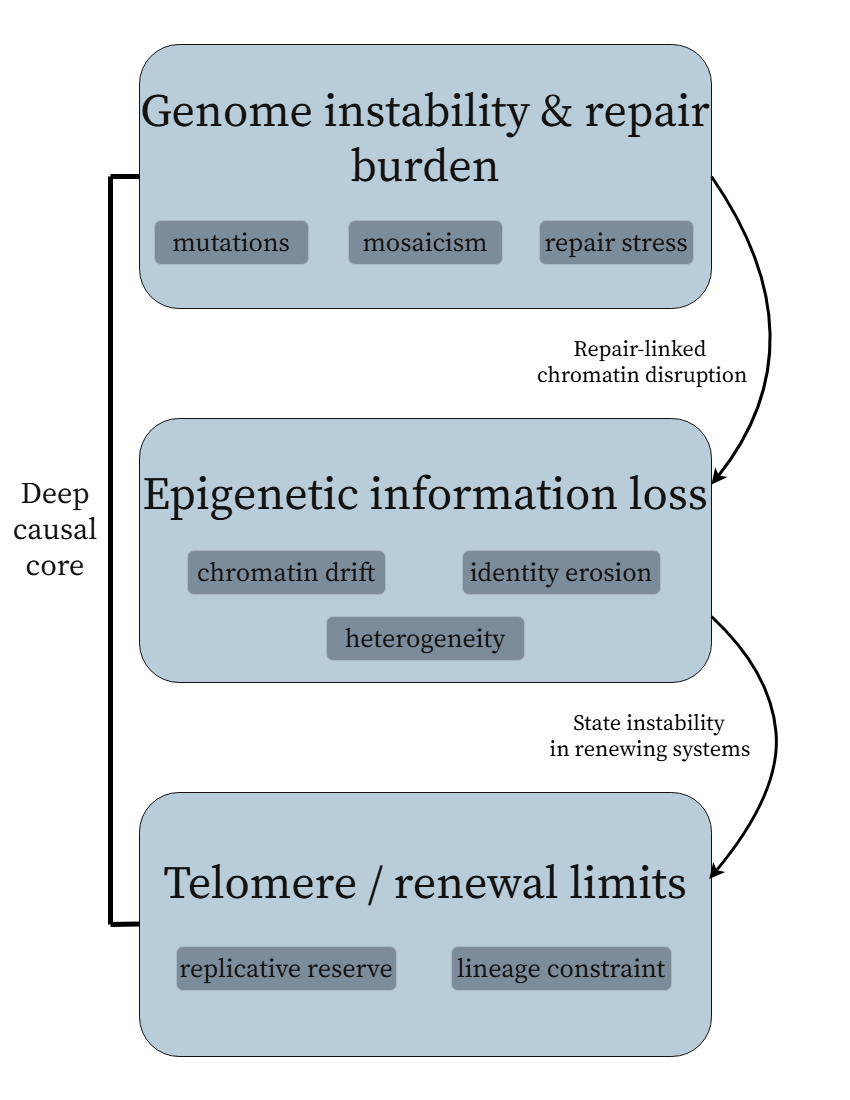
Figure 2. The deep causal core of the fidelity-loss model. Genome instability and repair burden generate somatic divergence and regulatory stress; epigenetic information loss converts that stress into erosion of cell-state fidelity; and telomere-linked renewal limits impose an additional lineage-specific boundary in renewing compartments.
The next layer in the hierarchy asks what happens when the systems charged with preserving macromolecular and organellar integrity begin to fail across the organism, regardless of whether cells are dividing or not.
Primary maintenance layer I: Proteostasis collapse, translational fidelity, and autophagy failure
If the preceding sections define the deepest pressures on aging as threats to genomic integrity, chromatin-state stability, and proliferative reserve, the next layer asks how cells preserve the functional machinery that actually executes biological life from moment to moment. The genome and epigenome may store information, but proteins carry out almost every immediate task of cellular existence: catalysis, structural support, trafficking, signaling, replication, transcription, repair, and degradation. Aging therefore cannot be understood only as damage to inherited information or instability of cell identity. It must also be understood as a failure to preserve the integrity of the expressed proteome. In this sense, proteostasis is not a side issue. It is the maintenance system that determines whether biological information, even when still largely present, can still be translated into coherent function [29,30].
The proteostasis network includes protein synthesis, folding, chaperone surveillance, trafficking, disaggregation, proteasomal degradation, lysosomal degradation, and autophagic turnover. Its importance to aging lies partly in universality: all animal cells depend on it, whether rapidly dividing or largely post-mitotic. But its theoretical importance lies in a more specific point. Proteostasis is the level at which upstream fidelity loss becomes biochemically immediate. A mutation may or may not alter function; an epigenetic shift may or may not destabilize a lineage. By contrast, a misfolded, mistranslated, inadequately cleared, or aggregation-prone proteome directly impairs the cell’s capacity to act. Proteostasis is therefore best placed not as the deepest origin of aging, but as the first major maintenance layer through which many upstream disturbances become phenotypically consequential [29,30].
The evidence that this layer fails early is one reason it deserves a high ranking. In C. elegans, collapse of proteostasis was shown to occur early in adulthood rather than only in late-life terminal decline [29]. This finding changed the field’s timing intuition. It suggested that age-related proteome instability is not merely accumulated debris left behind after other systems have already failed. Instead, maintenance of the proteome can begin to deteriorate surprisingly soon after reproductive maturity, long before catastrophic organismal dysfunction becomes obvious [29]. That timing is consistent with the broader logic of this paper: aging is not best understood as a sudden late collapse, but as an early and progressive weakening of the systems that preserve biological fidelity. Proteostasis is one of the clearest examples of that principle.
The conceptual consequence is important. If proteostasis fails early, then aging cells are likely operating for much of life with an increasingly corrupted expressed state even before overt pathology emerges. Proteins that misfold more readily, assemble less efficiently, or persist longer than they should can change signaling thresholds, stress responsiveness, metabolic flux, and organelle maintenance in ways that are subtle at first but cumulative over time. In that sense, proteostasis collapse can act as a multiplier of upstream damage: genomic or epigenetic imperfections need not be dramatic if the cell’s capacity to buffer their functional consequences is already weakening. This is why the proteostasis literature has often treated aging itself as an event of proteostasis collapse, not because protein homeostasis explains everything, but because it occupies a uniquely strategic position between information and function [30].
Autophagy is one of the clearest mechanistic arms of this maintenance layer. It is especially important because it couples proteome quality control to organelle turnover, metabolic adaptation, and stress resistance. In C. elegans, autophagy was required for lifespan extension by dietary restriction, demonstrating that a major longevity intervention depends on intact cellular recycling and clearance machinery [31]. This result is not easily dismissed as an epiphenomenon. It implies that cells cannot fully convert a pro-longevity systemic signal into extended lifespan unless they retain the capacity to remove damaged components and repurpose internal resources [31]. More recent review work makes clear that autophagy itself declines with age at multiple levels, including initiation, cargo recognition, membrane dynamics, lysosomal competence, and flux completion [32]. In other words, one of the cell’s core self-cleaning and renewal systems becomes less reliable with age precisely when the burden on it is increasing.
This duality—centrality to longevity and decline with age—makes autophagy particularly informative for theory-building. It suggests that aging is not only caused by the generation of damage but also by the deterioration of the mechanisms that would normally prevent that damage from becoming systemically important. A good theory must therefore distinguish insult from buffering capacity. The autophagy literature strongly supports the view that aging accelerates when the buffering layer weakens [31,32]. This fits naturally into the biological-fidelity framework: cells lose not only correct information, but also the ability to maintain clean execution of that information in the face of stress, turnover, and macromolecular wear.
The Hydra evidence makes the causal importance of this layer even harder to ignore. Hydra is valuable precisely because some Hydra species exhibit negligible senescence, making them a natural challenge to any claim that aging is an unavoidable direct consequence of animal existence [84]. In that context, the finding that defective autophagy in Hydra epithelial stem cells can drive an aging phenotype is extremely informative [85]. It shows that in a normally non-senescent or weakly senescent system, failure of a core maintenance process can be sufficient to unmask aging-like decline. This does not prove that autophagy is the universal root cause of aging. But it does show that sustained cellular maintenance, especially turnover of damaged proteins and organelles, is not a secondary luxury. It is one of the conditions under which low-senescence states can be preserved at all [85]. That gives autophagy unusual comparative weight within the hierarchy.
Proteostasis, however, is not only about clearing bad proteins after they arise. It also depends on making proteins correctly in the first place. Here translational fidelity becomes especially relevant. A 2022 study showed that mice with error-prone protein synthesis exhibit premature-aging phenotypes, arguing that increased proteome corruption at the point of translation can itself drive organismal decline [117]. More recent work reported that translational error increases with age in mice in an organ-dependent manner, suggesting that the accuracy of protein synthesis itself is not fixed across the lifespan [118]. These results deserve more attention than they usually receive because they extend the concept of biological fidelity into the proteome with unusual clarity. Proteins can age not only because they are damaged after synthesis, but because they are increasingly synthesized incorrectly. In effect, the expression of biological information itself becomes noisier.
This point is theoretically powerful because it parallels the logic of genome instability and epigenetic drift. At the DNA level, sequence can become inaccurate; at the chromatin level, interpretation can become inaccurate; at the proteome level, execution can become inaccurate. Translational error therefore fits naturally into a multiscale fidelity-loss model. It shows that aging is not confined to the storage of information but extends to the machinery that instantiates that information as working biology [117,118]. The implication is that proteostasis should be broadened from “maintenance of proteins after they exist” to “maintenance of accuracy across the full life cycle of proteins,” including synthesis, folding, deployment, and clearance.
At the same time, this layer is not purely monotonic. The broader stress-response literature reminds us that mild challenge can be adaptive. Concepts such as mitohormesis show that limited cellular stress can trigger protective programs that improve long-term resilience [37]. This matters here because it cautions against interpreting every perturbation of proteostasis as directly pro-aging. The issue is not that cells must never experience proteotoxic pressure. The issue is whether they can mount adaptive responses and restore homeostasis without progressively compromising fidelity. When stress responses remain effective, they may extend healthspan; when they become chronically activated, incomplete, or energetically unsustainable, proteostasis failure becomes part of the aging process [30,37]. This distinction mirrors one that will recur later with senescence and inflammation: responses that are initially adaptive can become pathogenic when persistence replaces resolution.
Proteostasis is also deeply coupled to other maintenance systems. Protein misfolding burdens mitochondria and the endoplasmic reticulum; defective mitochondrial function can worsen proteostatic stress; lysosomal insufficiency impairs autophagic turnover; metabolic depletion can compromise chaperone and degradation systems. Interventions such as NAD repletion, while often discussed under mitochondrial aging, also reinforce the broader idea that maintenance systems are interconnected rather than isolated [39]. The value of placing proteostasis here in the hierarchy is precisely that it explains how very different upstream insults can converge onto a common biochemical crisis: the cell increasingly loses the ability to keep its working parts accurate, clean, and replaceable.
For these reasons, proteostasis collapse, autophagy decline, and translational inaccuracy are best treated as a primary maintenance layer of aging. They are not the deepest origin of aging in the same sense as genome instability or epigenetic information loss, because they are strongly modulated by upstream rate-control pathways, metabolic context, and stress history [29–32]. But they are too central, too early, and too causal to be relegated to a late downstream consequence. In the present framework, they represent the first major point at which biological fidelity is lost not merely in stored information, but in the active molecular workforce of the cell. Aging, in this sense, includes progressive corruption of the proteome itself.
That conclusion prepares the next section directly. If proteostasis and autophagy define one major maintenance layer, the mitochondrial system defines another—one that is intimately coupled to protein quality control, metabolic adaptation, and the cell’s ability to sustain coordinated function under stress. The next question is therefore not whether mitochondria matter, but how they matter without collapsing the theory back into the older and overly simple free-radical model.
Primary maintenance layer II: Mitochondrial quality control, mito-nuclear uncoupling, and NAD decline
If proteostasis and autophagy define one major maintenance layer of aging, mitochondria define another. Few components of the cell sit at a more strategic junction between information, metabolism, stress response, and survival. Mitochondria are not merely ATP generators. They integrate redox state, apoptotic signaling, intermediary metabolism, metabolite availability for chromatin regulation, innate immune activation, and the quality-control pathways that determine whether damaged cellular components are repaired, recycled, or eliminated [33]. For that reason, mitochondrial decline cannot be treated as a narrow bioenergetic defect appended to aging after the fact. It is a central maintenance problem. At the same time, it must be framed carefully. The mitochondrial layer is clearly causal, but it is not well described by the older and overly simple view that aging is primarily a matter of oxidative damage steadily rising with time [33,36].
The strongest evidence for the importance of this layer comes from models in which mitochondrial genomic fidelity is directly compromised. In proofreading-deficient mitochondrial mutator mice, mtDNA deletions and clonal mutations drive premature-aging phenotypes, showing that mitochondrial genomic instability can be sufficient to accelerate organismal decline [34]. This is a crucial result because it moves mitochondrial dysfunction out of the purely correlational realm. It demonstrates that when one undermines the integrity of the mitochondrial genome, a broad aging-like phenotype can emerge. In a related line of work, increased mtDNA mutation burden in skeletal muscle induces mitochondrial dysfunction, apoptosis, and sarcopenia, linking mitochondrial genomic instability to one of the clearest and most clinically important phenotypes of mammalian aging [35]. Together, these findings establish that mitochondrial deterioration is not simply an accompaniment of aging tissues. Under the right conditions, it can act as a driver of age-like degeneration.
Yet the mutator models also reveal why the mitochondrial layer should not be collapsed into a free-radical clock. The mitochondrial free-radical theory of aging was historically attractive because it provided a simple mechanism: mitochondria produce reactive oxygen species, ROS damage mitochondria and other cellular components, and this self-reinforcing cycle drives aging. But subsequent work has made that picture too coarse to remain sufficient. Critical reviews have shown that the relationship between oxidative stress and aging is inconsistent across models, tissues, and interventions, and that ROS cannot be treated as a monotonic master variable [36]. The central problem is not that ROS are irrelevant; they clearly matter in signaling and damage. The problem is that mitochondrial aging cannot be reduced to ROS load alone. Mitochondria fail as organelles with genomes, proteomes, membranes, dynamics, trafficking, turnover pathways, and communication channels. Their role in aging is therefore richer and more system-like than the older oxidative-damage model allowed [33,36].
The concept of mitohormesis sharpens this point. Mild mitochondrial stress can activate protective responses that improve resilience and, in some contexts, extend lifespan [37]. This observation is fatal to any simplistic interpretation in which all mitochondrial perturbation is intrinsically pro-aging. Instead, it suggests that what matters is whether mitochondrial stress remains within a range that can be sensed, buffered, and adaptively resolved. When adaptive capacity remains high, mitochondrial perturbation may strengthen maintenance systems. When it becomes chronic, excessive, or poorly resolved, the same organellar stress can contribute to decline [37]. The implication for the present theory is straightforward: mitochondrial dysfunction is causally important, but its effects are nonlinear because mitochondria are embedded in broader stress-response and quality-control networks rather than functioning as isolated damage sources.
This is why mitochondrial quality control is the more appropriate framing. The relevant question is not simply how damaged mitochondria become, but whether cells can maintain mitochondrial fidelity through biogenesis, fusion-fission dynamics, proteome surveillance, mitophagy, metabolite exchange, and signaling to the nucleus. In this sense, the mitochondrial layer parallels the proteostasis layer described above. Aging may not arise because a single organellar lesion accumulates inexorably; it may arise because the systems that preserve mitochondrial competence and coordinate it with cellular needs become progressively less reliable [33]. Support for this view comes from hematopoietic stem-cell biology, where a mitochondrial unfolded-protein-response checkpoint helps regulate stem-cell aging [61]. That finding places mitochondrial stress surveillance directly inside a regenerative compartment central to organismal aging and underscores that mitochondrial failure is not just a passive consequence of tissue decline. It is part of the machinery that shapes stem-cell fate and long-term maintenance [61].
One of the most illuminating developments in this area has been the recognition that mitochondrial aging is often a problem of communication, not only damage. A striking example is age-related NAD decline. Declining NAD+ induces a pseudohypoxic state in which nuclear-mitochondrial communication becomes disrupted, in part through HIF-1α stabilization and impaired coordination between nuclear and mitochondrial programs [38]. This finding substantially reframes the problem. It suggests that mitochondrial aging is not adequately described by saying that old mitochondria simply make less energy or accumulate more injury. Rather, the coordination between nucleus and mitochondria—between genetic instruction and organellar execution—becomes progressively uncoupled [38]. In a theory centered on biological fidelity, this is exactly the kind of defect that deserves elevation: a failure not merely of a part, but of the relationship between parts.
The significance of NAD biology becomes even greater because the defect is at least partly reversible. NAD+ repletion improves mitochondrial function, enhances stem-cell performance, and extends lifespan in mice [39]. These outcomes imply that a substantial fraction of mitochondrial aging lies not in irrecoverable destruction of organelles, but in an altered regulatory and metabolic state that can be shifted toward a more youthful configuration. This does not mean mitochondrial aging is trivial or wholly plastic. It means that one of its central features is a degradation of coordination that can, within limits, be restored [38,39]. That observation fits the broader pattern emerging across the paper: some of the most important layers of aging are not best understood as accumulations of terminal damage, but as progressively destabilized systems of maintenance and communication.
The mitochondrial layer therefore occupies a distinctive position in the hierarchy. It is not as deep as genome instability in the sense of being the first source of heritable divergence across the soma, nor is it as directly explanatory of cell-state drift as epigenetic information loss. But it is among the most important maintenance layers because it determines whether cells can still transform stored biological information into usable energy, balanced signaling, controlled apoptosis, and metabolic homeostasis. Once mitochondrial quality control weakens, the consequences radiate outward: proteostasis becomes harder to sustain, redox signaling becomes less discriminating, inflammatory signaling can intensify, stem-cell function may decline, and stress adaptation becomes less reliable [33,38,39,61]. In this way, mitochondrial dysfunction acts less like a single lesion and more like a destabilization of cellular infrastructure.
This perspective also explains why mitochondrial dysfunction can look so variable across tissues and still belong to a common architecture. Tissues differ radically in energetic demand, mitophagic burden, mitochondrial density, regenerative strategy, and exposure to inflammatory or mechanical stress. Muscle, neurons, immune cells, and stem-cell compartments therefore experience mitochondrial decline differently. Yet the underlying problem may still be shared: the loss of reliable organelle turnover, organelle-genome integrity, and nucleus-organelle coordination [33–35,38,39]. In that sense, mitochondrial heterogeneity does not weaken its place in the theory. It shows how a common maintenance layer can generate divergent executor phenotypes depending on tissue context.
For these reasons, the mitochondrial layer is best treated as a quality-control and signaling layer embedded in a larger adaptive system. Its causal role is real, but it is not adequately captured by a simple damage metric. mtDNA instability can drive progeroid phenotypes and sarcopenia [34,35]; ROS-centric explanations are insufficient on their own [36]; mild mitochondrial stress can be beneficial through mitohormesis [37]; declining NAD+ can uncouple nuclear and mitochondrial programs [38]; and restoring NAD+ can partially reverse functional decline [39]. The unifying lesson is that mitochondria matter most in aging not as passive batteries that run down, but as dynamic organelles whose integrity, turnover, and communication with the rest of the cell must be continually maintained. Once that maintenance falters, fidelity is lost at one of the most operationally important levels of biology.
This framing also prepares the transition to the next section. If proteostasis and mitochondria are the two most important primary maintenance layers, the question becomes what controls the rate at which they are preserved or sacrificed over the life course. That question leads directly to the nutrient-sensing and life-history allocation pathways—systems that do not by themselves explain all aging, but that appear to govern how aggressively organisms invest in growth, reproduction, maintenance, and repair.
Rate-control layer: Nutrient sensing and life-history allocation
The preceding sections identified deep sources of aging pressure—genome instability, epigenetic information loss, renewal limits—and the maintenance systems that buffer them, especially proteostasis, autophagy, and mitochondrial quality control. But these layers alone do not explain one of the most striking facts in the field: the rate of aging is highly plastic. Organisms with similar basic cellular architecture can age at very different speeds, and lifespan can be profoundly altered by relatively small perturbations to conserved signaling pathways. This is why nutrient sensing deserves a distinct place in the hierarchy. IIS–mTOR–FOXO and related nutrient-responsive pathways are best understood not as the deepest origin of aging, but as a rate-control layer that determines how aggressively organisms invest in growth, reproduction, stress resistance, recycling, and maintenance over time [40–42,105–107].
The worm data first made this impossible to ignore. Mutation of daf-2 in C. elegans approximately doubled lifespan, demonstrating that aging rate is not fixed by passive wear alone but can be dramatically altered by a conserved endocrine-like signaling pathway [40]. The significance of that result is hard to overstate. It converted aging from something that looked largely inevitable into something that appeared biologically regulated. Just as importantly, it implied that the pace of decline is at least partly governed by cellular decisions about resource use, stress response, and maintenance investment, not only by the unavoidable production of damage [40]. In other words, the organism does not merely suffer aging; it participates in determining its tempo.
The fly literature confirmed that this was not a worm-specific anomaly. Loss of CHICO, the Drosophila insulin receptor substrate, extends lifespan [105]. Modulation of genes in the TOR pathway also extends lifespan in flies [106], and dFOXO regulates lifespan in brain and fat body, tying longevity to a transcriptional effector downstream of insulin-like signaling [107]. Taken together, these experiments show that the major nutrient-sensing axis is conserved across distinct animal lineages and that longevity can be altered at multiple nodes within the same general signaling architecture [105–107]. This convergence matters because it argues against the idea that lifespan extension in simple organisms is achieved by unrelated tricks. Instead, it suggests that animals possess deeply conserved systems for tuning the balance between anabolic growth and maintenance.
The mammalian and primate data make the same point in a more translationally relevant form. Rapamycin extended lifespan in mice even when treatment began late in life, implying that nutrient-sensing pathways do not merely establish developmental trajectories and then disappear from the aging process; they continue to regulate the pace of decline during adulthood and late life [41]. Caloric restriction improved health and survival in rhesus monkeys, reinforcing the argument that conserved metabolic and nutrient-response systems shape lifespan in primates as well [42]. In humans, FOXO3A genotype is strongly associated with longevity, which does not prove causation in the same way as the interventional animal data, but does place the same signaling axis within the human lifespan problem [108]. Across worm, fly, mouse, primate, and human evidence, the inference is consistent: aging rate is unusually sensitive to nutrient-sensing biology [40–42,105–108].
The key interpretive question is what these pathways are actually regulating. The most plausible reading is not that IIS–mTOR–FOXO by themselves “are aging,” but that they govern life-history allocation at the molecular level. They sense nutrient abundance, growth cues, and metabolic state, then modulate whether the organism behaves as though resources should be directed toward anabolic growth and reproduction or toward recycling, maintenance, and resistance to stress. This interpretation is supported by the fact that autophagy is required for lifespan extension by dietary restriction [31], and by the broader observation that adaptive stress responses such as mitohormesis can support longevity when properly engaged [37]. Nutrient-sensing pathways therefore appear to sit above several of the maintenance layers described earlier, controlling whether cells and tissues fund the very processes that preserve biological fidelity [31,37]. The same logic is consistent with work showing that metabolic restoration, including NAD-related interventions, can improve mitochondrial and stem-cell function [39]: the rate-control layer and the primary maintenance layers are distinct, but intimately coupled.
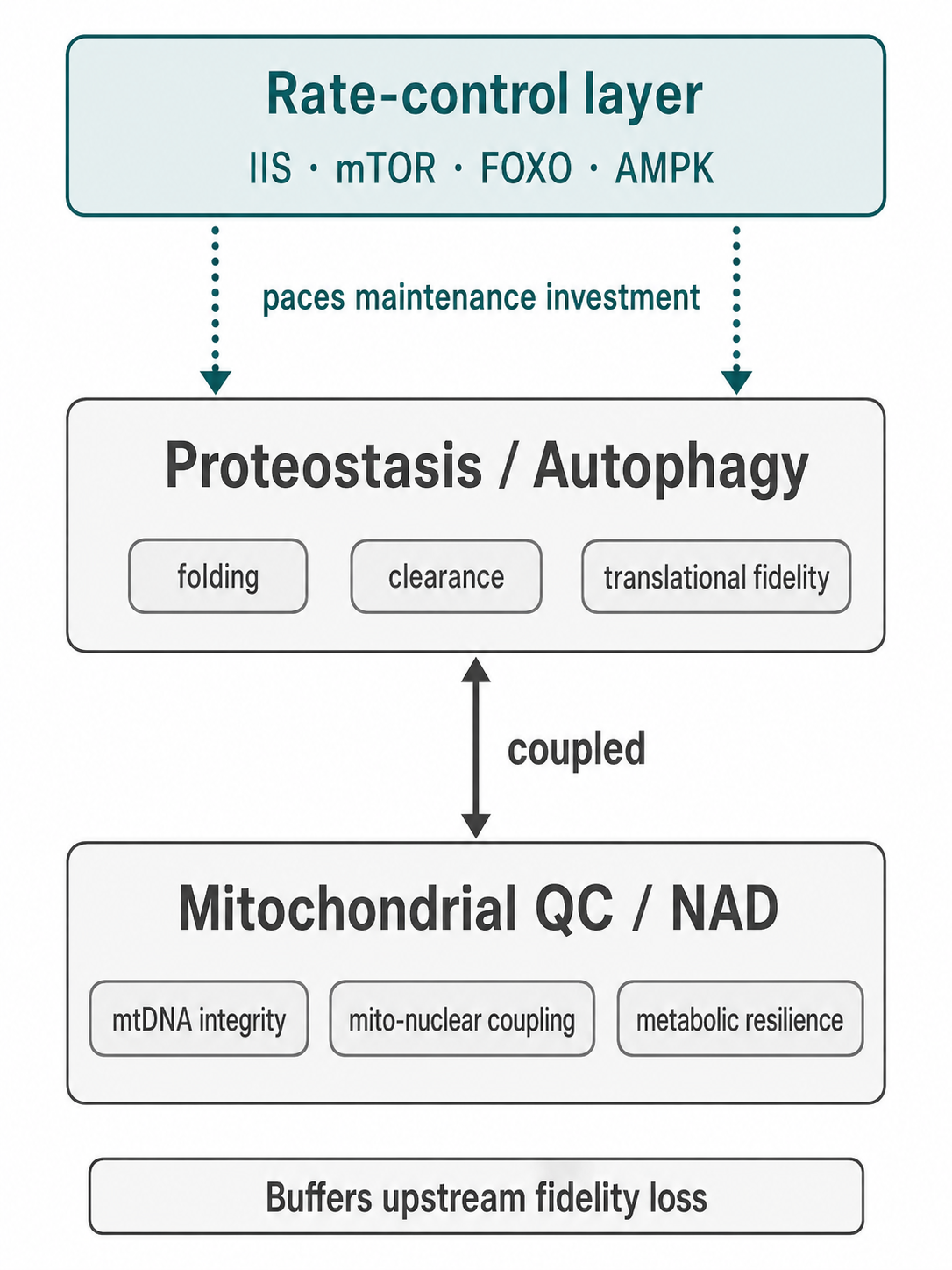
Figure 3. Primary maintenance systems and their rate control. Proteostasis/autophagy and mitochondrial quality control/NAD coupling act as the principal maintenance layers that buffer upstream fidelity loss, while conserved nutrient-sensing pathways regulate how strongly these systems are funded over the life course.
This placement also clarifies why nutrient sensing should not be mistaken for the deepest cause of aging. If these pathways were the sole origin of aging, then aging would look more like a centrally scripted program and less like the interaction of damage, imperfect maintenance, and adaptive response. The evidence does not support such a reduction. Deep lesions still matter; epigenetic and proteostatic failure still matter; mitochondria and stem cells still matter. What nutrient-sensing pathways appear to do is determine how quickly those liabilities accumulate and how robustly they are buffered. They are therefore better viewed as controllers of pace than as the singular source of decline. Their power comes from being upstream of many maintenance decisions, not from replacing the lower layers of the hierarchy.
This distinction is especially important for a multiscale theory of aging. A rate-control layer explains how the same fundamental vulnerabilities can produce different lifespans across species, individuals, and conditions without requiring wholly different aging mechanisms in each case. Organisms do not need different kinds of biology to age faster or slower; they can age at different speeds because conserved signaling systems tune the expenditure devoted to maintenance and repair versus growth and immediate reproductive economy. In this view, nutrient sensing is the molecular face of a broader biological tradeoff: the preservation of fidelity is costly, and organisms vary in how strongly they pay that cost over time.
Placed in the hierarchy this way, nutrient sensing links ecology and life history to cellular aging. Deep causal layers define what can go wrong; maintenance layers determine whether cells can buffer those problems; nutrient-sensing pathways regulate how much of the organism’s resources are allocated to buffering them in the first place. This framing also prepares the next step of the argument. If nutrient sensing sets the pace at which fidelity is lost, the next question is how cells respond once damage and maintenance failure cross a threshold that cannot simply be buffered away. That question leads directly to senescence: a protective response that initially contains harm, but later becomes one of the strongest conversion mechanisms from local damage to organismal aging.
| Mechanism group | Role in the early hierarchy |
|---|---|
| Genome instability | Creates mutation burden, repair burden, and somatic selection pressure. |
| Epigenetic / chromatin drift | Erodes cell-state fidelity and increases transcriptional mis-specification. |
| Telomere / renewal limits | Constrain high-turnover compartments and stem-cell reserve. |
| Proteostasis / autophagy | Maintain folding, clearance, recycling, and macromolecular quality. |
| Mitochondrial QC / NAD | Maintain energy balance, mito-nuclear coupling, and metabolic resilience. |
| Nutrient sensing | Regulates maintenance investment and the tempo of fidelity loss. |
Table 2. Early hierarchy of fidelity loss and maintenance buffering. Deep causal layers create the initial pressures on biological fidelity, while primary maintenance systems and rate-control programs determine how quickly those pressures accumulate.
Antagonistic response layer: Senescence as protection that becomes pathology
Among the mechanisms that shape aging, cellular senescence is unusual because it is most intelligible not as a primary source of damage, but as a response to damage, excessive mitogenic stress, and failures of cellular fidelity that can no longer be safely buffered. The classical Hayflick observations established that human diploid cells have a limited proliferative lifetime, introducing the phenomenon now recognized as replicative senescence [43]. Subsequent work broadened this concept: senescence is now understood as a durable stress response triggered by multiple insults, including aging, genomic or epigenomic damage, oncogenic signaling, and tissue injury, in which cells withdraw from proliferation and adopt a distinct secretory and regulatory state [43,102]. In the architecture proposed here, senescence therefore belongs in an antagonistic-response layer. It is what cells do when continued division or continued normal function would become more dangerous than arrest.
This placement immediately clarifies why senescence should not be treated as the root cause of aging. At least in its initial form, senescence is protective. It halts the growth of cells under potentially oncogenic or otherwise destabilizing conditions and thereby functions as a barrier to malignant transformation and uncontrolled propagation of damaged states [102]. The same basic logic explains why senescence can appear in contexts that are not simply degenerative. In cutaneous wound healing, senescent fibroblasts and endothelial cells arise early after injury and accelerate wound closure by promoting myofibroblast differentiation through secretion of PDGF-AA; recombinant PDGF-AA can rescue the delayed closure seen in senescence-free wounds [102]. These results are important because they demonstrate that senescence is not inherently pathological. In the right temporal and tissue context, it is part of normal tissue repair.
The problem emerges when senescence persists. Once senescent cells accumulate chronically rather than transiently, the same response that once limited harm begins to generate it. This shift has some of the strongest causal evidence in the modern aging field. In the BubR1 progeroid mouse background, inducible removal of p16^Ink4a-positive senescent cells delayed the onset of age-related pathologies in adipose tissue, skeletal muscle, and eye, while even late-life clearance attenuated progression of already established disorders [44]. This was followed by evidence in wild-type mice showing that naturally occurring p16^Ink4a-positive cells that accumulate during adulthood shorten healthy lifespan, and that their clearance delays tumorigenesis and preserves function in multiple organs [45]. These studies together make an unusually strong point: senescent cells are not merely markers of damaged tissue. Once retained, they become active contributors to age-related dysfunction.
The translational significance of that conclusion was strengthened by senolytic studies. Intermittent administration of senolytics reduced naturally occurring senescent-cell burden, alleviated physical dysfunction, and increased post-treatment survival in naturally aged mice, while also counteracting dysfunction induced by transplantation of senescent cells [46]. That result does not imply that all aging is reducible to senescence, nor that every benefit of senolysis is necessarily specific to one senescent-cell subtype. But it does show that chronic senescent-cell burden is therapeutically actionable and that reducing it can improve late-life function. This is exactly what one would expect if senescence occupies a conversion layer between upstream molecular damage and downstream organismal decline.
What makes senescence especially powerful as a driver of late-life pathology is that it is not only a cell-autonomous arrest state. Through the senescence-associated secretory phenotype, or SASP, senescent cells influence their neighbors and the surrounding tissue environment. Human cells induced to senesce by genotoxic stress secrete numerous inflammatory and pro-remodeling factors, and this secretory phenotype is amplified by oncogenic RAS or loss of p53 function [101]. The SASP therefore converts a local proliferative brake into a cell-nonautonomous signaling state that can alter tissue structure, recruit immune responses, modify extracellular context, and in some settings promote malignant or degenerative phenotypes nearby [101]. This is the key reason senescence belongs higher than a simple endpoint in the hierarchy: it is one of the mechanisms by which local failures are broadcast outward into tissue-level dysfunction.
The persistence of senescent cells with age also requires explanation, and recent work provides one. Senescent cells upregulate the immune checkpoint molecule PD-L1, which drives immune-cell inactivation; this induction depends on the proinflammatory program, can be triggered in neighboring nonsenescent cells by secreted factors, and is observed in naturally aged tissues [56]. This suggests that senescent cells are not only inflammatory broadcasters but can also become more difficult for the immune system to clear. In other words, the senescent state contains within it a mechanism of persistence: a cell population that should ideally appear transiently and then be removed can instead entrench itself. That feature helps explain why senescence, initially protective, becomes increasingly pathological with age.
Taken together, the evidence supports a precise placement of senescence in the aging hierarchy. Senescence is not best understood as the earliest cause of aging, because it is usually induced by upstream problems—DNA damage, telomere dysfunction, oncogenic or metabolic stress, chromatin instability, or tissue injury. Nor is it best understood as a passive endpoint, because chronic senescent-cell burden clearly drives pathology and can be therapeutically reduced [44–46]. Rather, senescence is a conversion mechanism: a protective response that initially contains damage and limits malignant risk, but which later becomes a potent engine of tissue dysfunction through persistence, SASP-mediated signaling, and incomplete immune clearance [44–46,101,102,56]. This is why senescence belongs at the transition point between deep fidelity loss and the amplifier loops that dominate later aging. The next section follows that logic directly by asking how chronic inflammation, innate danger sensing, and retrotransposon derepression transform diverse local lesions into a more unified organismal aging syndrome.
Amplifier layer I: Inflammaging, innate danger sensing, and retrotransposon derepression
Once senescence, mitochondrial dysfunction, chromatin instability, proteostatic burden, and tissue damage begin to accumulate, aging enters a phase in which local failures are no longer merely local. The most important bridge by which they become organism-wide is chronic inflammation. This is why inflammaging belongs high in the amplifier tier of the present hierarchy. It is not best understood as the first cause of aging, because many different upstream insults can trigger it. But it is one of the strongest mechanisms by which heterogeneous cellular failures are converted into a recognizable common syndrome of late-life decline [47–49]. In this sense, chronic inflammation is less an isolated hallmark than a convergence state.
The importance of that distinction is easy to miss. Inflammation is often described either too narrowly, as though it were just another damaging process, or too broadly, as though it were the singular root cause of aging. The evidence supports neither simplification. Chronic low-grade inflammation is better understood as a systemic amplifier that integrates multiple sources of cellular distress: genomic instability, mitochondrial dysfunction, extracellular debris, tissue injury, microbial products, persistent senescent cells, and failures of immune resolution [47,48]. It is therefore central not because it explains where aging starts, but because it explains how many distinct forms of cellular fidelity loss begin to look similar at the level of the organism. Once persistent inflammatory tone is established, tissues no longer age in isolation; they age in an increasingly shared inflammatory environment.
This is also why the link between inflammaging and immunosenescence is so important. Aging does not simply produce more inflammatory stimuli; it also alters the immune system’s ability to distinguish, contain, and resolve them [49]. The result is not merely heightened activation, but a shift in immune set point. Older organisms tend to show both impaired host defense and exaggerated low-grade inflammatory signaling, meaning that immune aging is simultaneously a failure of surveillance and a failure of restraint [49]. That duality fits the broader theory well. It suggests that late-life inflammation persists not only because more damaged material exists, but because the systems charged with clearing it and returning tissues to baseline no longer do so efficiently.
The innate danger-sensing pathways cGAS–STING and NLRP3 are especially informative because they make the conversion from local cellular failure to systemic inflammation mechanistically explicit. cGAS–STING responds to misplaced self-DNA, including cytosolic DNA generated by nuclear instability, mitochondrial damage, micronuclei, or incomplete clearance of damaged chromatin. A recent study showed that cGAS–STING signaling can actively drive aging-related inflammation and neurodegeneration in mice, directly linking intracellular damage sensing to organismal age pathology [50]. This matters enormously for the present theory. It means that DNA damage does not remain confined to the genome-repair layer. Under conditions of incomplete containment, the products of genomic instability become interpreted as immune danger signals. Damage is thus translated into inflammation.
NLRP3 adds a complementary logic. Rather than reading DNA specifically, it functions as a broader inflammatory execution node that can be activated by a wide range of danger-associated inputs, including mitochondrial dysfunction, lysosomal stress, altered metabolites, extracellular crystals, and cellular debris [51]. In the aging context, this is exactly the sort of architecture one would expect from a late amplifier. NLRP3 is not a singular origin but a convergence device. It allows many different upstream perturbations to feed into a shared inflammatory output [51]. Put differently, cGAS–STING and NLRP3 explain why aging can remain mechanistically heterogeneous at the molecular level yet become phenomenologically coherent at the organism level. They translate diversity of insult into commonality of response.
This conversion becomes even more striking when chromatin failure and retrotransposon derepression are added to the picture. LINE-1 elements, normally repressed in youthful cells, can become activated in senescence and aging-associated states [53–55]. In senescent cells, LINE-1 activation can drive type I interferon responses, indicating that failure to silence retrotransposons is sufficient to generate innate immune signaling [53]. SIRT6 helps repress LINE-1 retrotransposons under normal conditions [54], but this repression fails with age and stress. In aged and SIRT6-deficient mice, LINE-1 derepression triggers cGAS-dependent inflammation and can be partially mitigated by nucleoside reverse-transcriptase inhibitors [55]. These findings are particularly important because they link an upstream epigenetic problem—loss of chromatin repression—to a downstream inflammatory amplifier. In the framework advanced here, this is exactly the kind of cross-layer coupling that a unifying theory must explain.
Retrotransposon biology is therefore not an isolated curiosity. It is a mechanistic bridge between deep and late layers of the hierarchy. Earlier sections argued that aging may involve loss of biological information at the chromatin level. The LINE-1 evidence shows one concrete route by which such loss can become systemically consequential: chromatin failure leads to genomic noise; genomic noise is sensed as danger; danger sensing drives inflammatory pathology [53–55]. This sequence is especially persuasive because it does not require a large increase in classical exogenous infection or trauma. Endogenous disorder becomes sufficient to trigger immune activation. Aging tissues, in effect, begin to interpret themselves as injured.
Senescent cells intensify this process further. Through the SASP, senescent cells are already known to secrete inflammatory and tissue-remodeling factors that alter local and systemic environments [101]. What Section 10 established was that senescence converts damage into a persistent secretory state. The present section adds that this state can be extraordinarily difficult to resolve. Senescent cells upregulate PD-L1, which promotes immune-cell inactivation and impairs their own clearance [56]. This matters because it gives inflammaging a persistence mechanism. The same cells that generate inflammatory tone and local damage signals can also suppress the surveillance systems that would otherwise remove them. Thus, chronic inflammation is not simply “more immune signaling.” It is a failure of the full cycle of injury response: detection occurs, but resolution and clearance become incomplete.
The distinction between transient and chronic responses is once again decisive. Senescence can be beneficial in wound repair [102], and immune activation is indispensable for host defense and tissue healing. The pathology of aging emerges when these programs lose temporal discipline. A response that should be acute becomes background. A signal that should recruit cleanup becomes a constant tone. A state that should resolve becomes self-maintaining. This is why chronic inflammation is such a powerful amplifier: it recruits the organism’s most system-wide response systems and then prevents them from returning to baseline. The result is a tissue environment that favors further dysfunction, further senescence, further immune dysregulation, and often further clonal selection.
In this way, inflammaging helps explain why so many late-life conditions share overlapping biology even when their initiating lesions differ. Whether the upstream insult is DNA damage, mitochondrial distress, proteostatic failure, dysbiosis, extracellular matrix remodeling, or stem-cell exhaustion, the inflammatory layer provides a mechanism by which those disturbances can be generalized and synchronized [47–49]. This does not mean inflammation is nonspecific noise. It means it is a common language into which many different aging insults are translated. That translation is one reason aging appears coordinated at the whole-organism level even when it originates in many local and heterogeneous processes.
Placed in the hierarchy, then, inflammaging is best understood as the main bridge by which deep cellular fidelity loss becomes organismal aging syndrome. cGAS–STING and NLRP3 connect intracellular disorder to innate immune output [50,51]. LINE-1 derepression connects chromatin failure to interferon and cGAS signaling [53–55]. Senescent cells broadcast inflammatory signals and evade immune removal through PD-L1-mediated mechanisms [56,101]. Immunosenescence weakens the system’s ability to clear and resolve the disturbances it detects [49]. Together, these features make inflammation not the beginning of the story, but one of the most powerful ways the story becomes self-amplifying.
This conclusion leads directly to the next amplifier layer. Once chronic inflammation is established, one of the tissues most strongly remodeled by it is the hematopoietic system itself. Aging blood and immune compartments do not merely reflect inflammatory state; they can become selective environments in which mutant or lineage-biased clones expand. The next section therefore examines clonal selection and hematopoietic skew as an amplifier mechanism through which somatic evolution becomes systemic aging.
Amplifier layer II: Clonal selection and hematopoietic skew
If chronic inflammation provides a common language through which many local lesions become systemic, clonal selection in the hematopoietic system provides one of the clearest mechanisms by which that language is stabilized, amplified, and distributed throughout the organism. This is why somatic evolution must be treated as more than an incidental accompaniment of aging. Clonal expansion in normal tissues is increasingly recognized as a general feature of aging rather than a phenomenon confined to overt cancer [10]. But the hematopoietic system is a particularly consequential case, because even a modest change in stem-cell composition can reshape the output of circulating immune and inflammatory cells across the entire body. In the present framework, hematopoietic clonal selection is therefore not placed among the deepest origins of aging; it is placed among the strongest amplifier mechanisms by which upstream damage becomes organism-wide pathology.
The starting point for this argument is age-related clonal hematopoiesis itself. Somatic clones carrying mutations in hematopoietic stem and progenitor cells become increasingly common with age, even in individuals without diagnosed hematologic malignancy, and their presence is associated with increased risk of blood cancer, coronary heart disease, ischemic stroke, and all-cause mortality [11]. This finding was a conceptual turning point because it demonstrated that clonal evolution in the blood is not a rare oncologic edge case. It is a widespread aging phenomenon with consequences that extend beyond the hematopoietic compartment. A clone need not become leukemia to matter. It is enough that it changes the inflammatory behavior, lineage output, or tissue interactions of the cells it produces [11].
This is what makes hematopoiesis different from many other aging tissues. In most organs, clonal expansion can remodel the local tissue environment; in blood, it can remodel the organism. Hematopoietic stem cells generate lineages that traffic through vasculature, populate peripheral tissues, participate in injury response, and regulate inflammation, immunity, and repair. A mutation that shifts the behavior of these cells is therefore not confined to the marrow niche. It becomes a whole-body variable. For this reason, clonal hematopoiesis is best understood as a privileged amplifier layer: it links the deep lesion-generating processes described earlier to system-wide inflammatory and degenerative consequences.
The TET2 story provides one of the strongest direct demonstrations of that logic. TET2-deficient clonal hematopoiesis accelerates atherosclerosis in mice, not merely by increasing clone size, but by altering macrophage behavior toward a more pro-inflammatory state [12]. This is the critical point. The pathogenic effect of the clone is not exhausted by its existence; it lies in the altered functional program of its descendants. A somatic mutation in a stem cell becomes vascular pathology because the myeloid cells derived from that stem cell now execute different inflammatory programs [12]. In other words, clonal hematopoiesis transforms genomic divergence into altered immune physiology. That is exactly the kind of transition expected of an amplifier layer.
The relationship is also bidirectional. Inflammatory environments can favor the expansion of Tet2-mutant clones, meaning that inflammation is not only a consequence of clonal hematopoiesis but also part of the selective landscape that promotes it [13]. This observation is crucial for causal ordering. It shows that hematopoietic clonal evolution does not sit outside the aging network as a parallel process. It is embedded in a feedback loop. Deep fidelity loss and chronic inflammatory signaling create conditions in which certain clones are preferentially selected; those clones in turn intensify inflammatory tone and age-related disease risk. The organism therefore enters a self-reinforcing state in which the output of the hematopoietic system progressively shifts toward phenotypes that worsen the very milieu that selected them.
This loop helps explain why hematopoietic aging so often presents as lineage skew rather than only simple stem-cell depletion. Aging hematopoiesis is characterized by increased myeloid bias, reduced lymphoid competence, and altered regenerative balance, suggesting that the problem is not merely fewer stem cells or more damage, but a change in which stem-cell states and lineages dominate [63]. Inflammatory signaling and niche remodeling are increasingly implicated in this process, meaning that lineage bias should be understood as both a product of selective pressures and a producer of downstream dysfunction [63]. This is why lineage skew is so important theoretically: it is the visible phenotype of somatic evolution acting within an aging tissue ecology.
Recent intervention work sharpens this further. Depleting myeloid-biased hematopoietic stem cells can rejuvenate the aged immune system, implying that age-associated lineage bias is not merely descriptive but functionally causal and at least partly reversible [62]. That result is important for two reasons. First, it supports the idea that aged hematopoiesis is actively maintained by a distorted clonal composition rather than simply exhausted in a terminal way. Second, it shows that correcting the composition of the stem-cell pool can improve systemic immune function, again underscoring that the hematopoietic compartment is not just another aging tissue. It is a central transmitter of organismal aging state [62].
The connection to inflammaging is therefore tighter than a simple “inflammation plus mutations” picture would suggest. Inflammation changes the competitive environment of hematopoietic clones [13,63]. Clonal hematopoiesis changes the inflammatory output of circulating immune cells [11,12]. Inflammasome-centered cytokine networks provide one plausible execution route for this amplification, since inflammatory myeloid programs can drive chronic tissue damage and age-related disease [12,51]. The hematopoietic system thus becomes both sensor and broadcaster: it registers upstream aging pressures through clonal selection and lineage skew, and it broadcasts those changes back into the organism as altered immunity, altered repair, and persistent inflammatory tone.
This placement also explains why clonal hematopoiesis is especially important in humans and other long-lived vertebrates. In short-lived organisms, aging may be dominated more directly by rapid catastrophic failures or simpler life-history constraints. But in long-lived animals with extensive tissue renewal, persistent immune surveillance, and prolonged exposure to inflammatory and metabolic stress, even subtle shifts in hematopoietic composition can accumulate enormous systemic consequences over time. The longer the organism lives, the more opportunity exists for somatic evolution in blood to interact with chronic inflammation, vascular injury, infection history, and tissue repair demands. Clonal hematopoiesis is thus one of the clearest examples of how longevity itself can make amplifier layers more important.
In the hierarchy proposed here, then, hematopoietic clonal selection and lineage skew are not the beginning of aging, but they are among the clearest ways aging becomes self-amplifying. Genome instability seeds somatic variants [10,11]. Inflammation and niche change select among them [13,63]. Selected clones alter immune and myeloid output, increasing chronic inflammatory and degenerative burden [12,51]. The resulting milieu further reshapes the clonal landscape. This is not merely a pre-cancer story. It is a general model of how somatic evolution in a mobile, system-wide tissue can propagate aging across the organism.
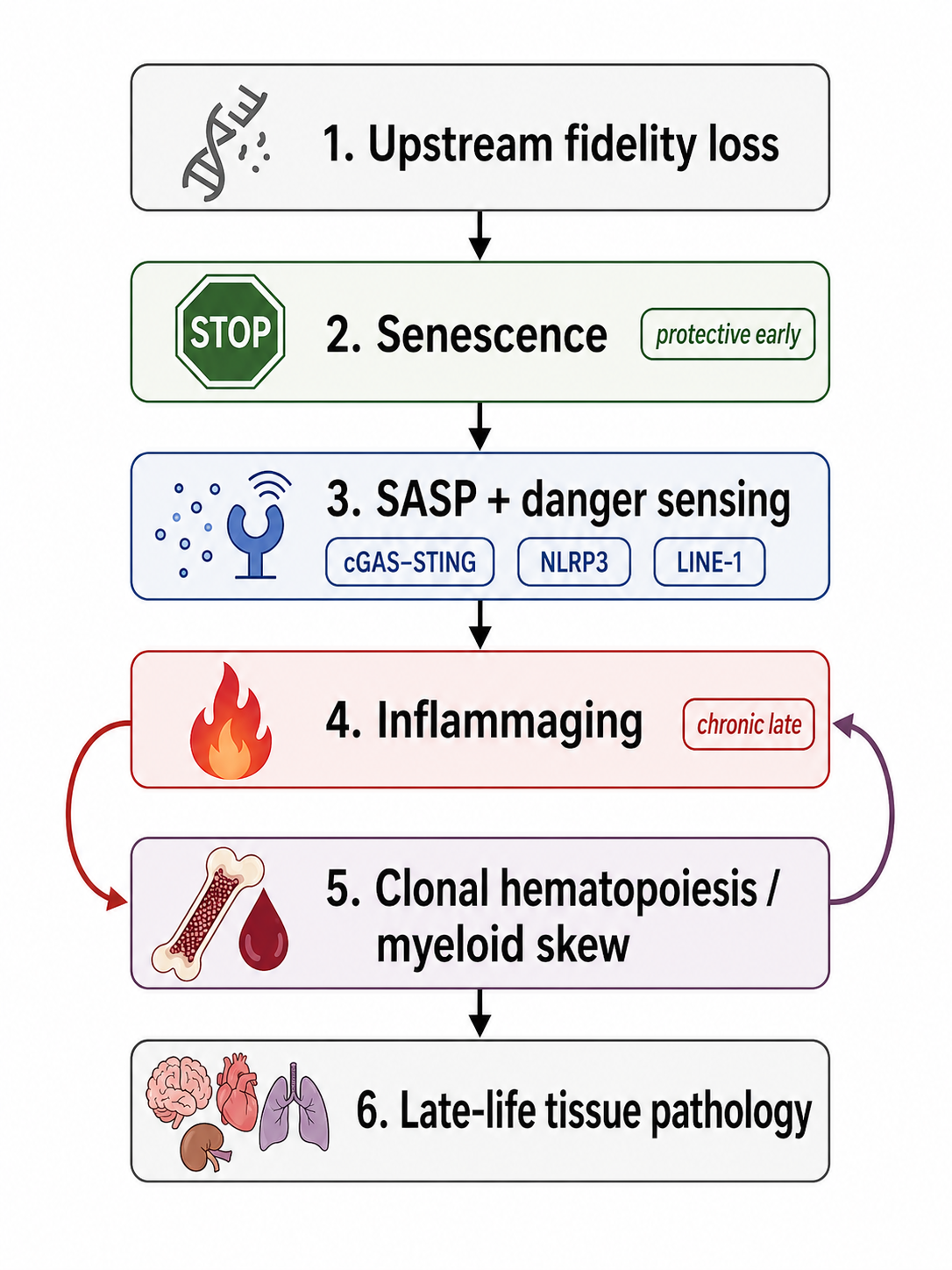
Figure 4. From protective response to chronic pathology. Senescence initially restrains damaged cells, but persistent SASP, innate danger sensing, chronic inflammation, and clonal hematopoietic remodeling convert local failures into self-reinforcing late-life pathology.
The broader theoretical lesson is that stem-cell lineage bias is both a driver and a reflection of aging. It reflects the long history of damage, repair, inflammation, and niche remodeling that has acted on the hematopoietic system. But it also drives future aging by altering what kinds of cells the organism produces in response to stress, injury, and ordinary tissue turnover. In that sense, the hematopoietic compartment provides one of the strongest examples of the logic underlying this entire paper: local losses of biological fidelity matter most when they are translated into altered system behavior. The next section extends that logic beyond blood and inflammation alone by examining how circulating factors, neuroendocrine programs, and organism-wide proteomic states synchronize local failures into a more coherent whole-organism aging phenotype.
Amplifier layer III: Systemic milieu, neuroendocrine coordination, and circulating state
By the point reached in the hierarchy so far, aging is no longer a purely local process. Deep fidelity loss has begun to generate chronic responses; senescent cells and inflammatory signaling have made tissue environments more hostile; clonal skew in hematopoiesis has started to alter the composition of circulating immune output. The next question is therefore unavoidable: how do these distributed failures begin to behave like a whole-organism state rather than a scattered collection of tissue problems? The answer suggested by the literature is that aging is coordinated, at least in part, by a cell-nonautonomous layer composed of circulating factors, central neuroendocrine control, and organism-wide state signals that propagate and synchronize local deterioration. This view is consistent with the broader systems picture emerging from single-cell and multi-omic aging studies, which show that aging is coordinated across tissues yet strongly heterogeneous in how it manifests locally [96,100].
The strongest early evidence for such coordination came from heterochronic systemic-exposure experiments. Exposure of aged progenitor cells to a young systemic environment rejuvenated their function, demonstrating that at least part of age-related decline in regenerative competence is not irreversibly fixed within the old cells themselves [57]. The principle extends beyond stem and progenitor compartments. In aged mice, exposure to young blood reversed pre-existing brain-aging phenotypes at molecular, structural, functional, and cognitive levels; heterochronic parabiosis induced synaptic plasticity-related transcriptional changes in the aged hippocampus, increased dendritic spine density, improved hippocampal synaptic plasticity, and young plasma administration improved contextual fear conditioning and spatial learning and memory [104]. These findings are among the most important in the field because they show that age state is at least partly transmissible through the systemic milieu. Old tissues are not merely old because of what has happened inside them; they are also old because of the environment in which they are embedded.
The hypothalamus provides a complementary line of evidence that aging is centrally coordinated as well as peripherally broadcast. In mice, hypothalamic IKK-β/NF-κB signaling has been shown to have a programmatic role in whole-body aging: preventing age-related hypothalamic or brain IKK-β/NF-κB activation retarded aging and extended lifespan, while mechanistic studies linked this pathway to suppression of gonadotropin-releasing hormone and age-related decline in hypothalamic GnRH; GnRH treatment partly restored neurogenesis and decelerated aging [64]. This does not mean aging is simply “caused by the hypothalamus.” Rather, it suggests that the hypothalamus functions as a central integrator that can convert inflammatory and metabolic information into organism-wide endocrine and neurogenic consequences. In the framework of this paper, that makes central neuroendocrine regulation an amplifier layer: it does not replace deeper causes, but it can shape how widely and how coherently they are expressed.
The hypothalamic stem-cell literature strengthens that interpretation. Hypothalamic stem/progenitor cells that co-express Sox2 and Bmi1 decline substantially with age in mice, and experimental ablation of these cells accelerates physiological aging and shortens lifespan. Conversely, implantation of healthy hypothalamic stem/progenitor cells into mid-aged mice retards aging and extends lifespan, and the work further showed that these cells contribute substantially to exosomal microRNAs in the cerebrospinal fluid, which themselves decline with age; central treatment with exosomes secreted by healthy hypothalamic stem/progenitor cells slows aging [65]. The significance of this finding goes beyond one brain region. It implies that organismal aging speed can be modulated by a central signaling node whose influence is exerted partly through diffusible information carriers. Aging is therefore not only a failure of local maintenance; it is also a failure of long-range coordination.
More recent work on hypothalamic Menin adds a concrete molecular regulator to this picture. Menin signaling in the hypothalamus declines with age and correlates with systemic aging and cognitive deficits; restoring Menin expression in the ventromedial hypothalamus of aged mice extended lifespan, improved learning and memory, and ameliorated aging biomarkers, whereas inhibiting Menin in middle-aged mice induced premature aging and accelerated cognitive decline [66]. Mechanistically, Menin was linked to neuroinflammatory and metabolic pathways, including D-serine metabolism. What matters most for the present theory is not the exclusivity of Menin as a master regulator, but what it demonstrates: central regulatory nodes can influence systemic aging through neuroinflammatory-metabolic integration and neural-circuit outputs. That makes the hypothalamus less a singular clock than a coordinating hub that can either dampen or accelerate organism-wide amplification of age-related dysfunction.
The plasma proteome literature shows that this organism-wide coordination is not only experimentally manipulable; it is also measurable. In a large study of 2,925 plasma proteins across 4,263 individuals aged 18–95 years, the human plasma proteome showed marked nonlinear changes across the lifespan, with distinct waves in the fourth, seventh, and eighth decades of life [103]. More recent work used organ-enriched plasma proteins and machine-learning models to estimate molecular age in 11 major organs across 5,676 adults in five cohorts and found that nearly 20% of the population had strongly accelerated aging in one organ and 1.7% were multi-organ agers. Accelerated organ aging conferred 20–50% higher mortality risk, accelerated heart aging was associated with roughly 250% increased heart-failure risk, and accelerated brain and vascular aging predicted Alzheimer’s disease progression [114]. These findings are central to the present section because they imply that the circulation carries structured information about both whole-body aging and organ-specific aging. Blood is therefore not just a convenient sampling compartment. It is a medium in which systemic aging state is represented and, plausibly, in part enacted.
This logic is extended by newer proteomic aging models. A proteomic age clock developed in the UK Biobank from 2,897 plasma proteins identified 204 proteins that predicted chronological age with high accuracy and showed that proteomic aging was associated with incidence of 18 major chronic diseases, multimorbidity, and all-cause mortality; the contributing proteins spanned extracellular matrix interactions, immune response and inflammation, hormone regulation, neuronal structure and function, and developmental pathways [115]. Organ-specific proteomic aging clocks have taken this further, showing that organismal and organ-specific plasma clocks predict disease onset, progression, and mortality beyond conventional clinical and genetic risk factors, with distinct organ clocks linked to distinct pathogenic pathways such as synaptic loss, vascular dysfunction, and glial activation in cognitive decline and dementia [120]. More recent plasma protein-based organ-specific aging and mortality models extend the same framework by explicitly treating chronic diseases as accelerated aging of specific organs or systems [121]. Collectively, these results argue that circulating state is not merely a biomarker layer placed on top of aging. It is part of the machinery through which local dysfunction becomes coordinated at the scale of organs and the organism.
These observations allow a more precise placement of this layer in the hierarchy. Systemic milieu and neuroendocrine coordination are not likely to be the deepest origin of aging, because the signals they coordinate are themselves shaped by deeper processes—genome instability, epigenetic drift, proteostatic decline, mitochondrial dysfunction, senescence, inflammation, and clonal remodeling. But they are far too powerful to be dismissed as passive readouts. Young blood can rejuvenate old tissues [57,104]; hypothalamic inflammatory and stem-cell programs can alter organismal aging rate [64,65]; hypothalamic Menin can modulate lifespan and cognition [66]; and plasma proteomic states capture coordinated organ-level aging and mortality risk [103,114,115,120,121]. The best interpretation is therefore that systemic milieu and central neuroendocrine signaling constitute an amplifier-coordinator layer: they propagate local failures, impose cross-tissue synchrony, and help determine whether aging remains compartmentalized or becomes organism-wide.
This placement also clarifies the transition to the next section. If blood and hypothalamic circuits help synchronize and distribute aging state, then other distributed ecological layers should matter as well—especially the microbiome, which can influence inflammatory tone, metabolite supply, barrier integrity, and regenerative context. The next amplifier layer therefore asks how host–microbe coupling tunes the organism-wide aging environment inside the larger architecture already described.
Amplifier layer IV: Microbiome and host–microbe coupling
If the previous section established that aging behaves in part like a circulating and neuroendocrine state, the microbiome adds a further layer of distributed coordination: the aging organism is not only regulating itself, it is regulating and being regulated by a large ecological system at its epithelial interfaces. In mammals especially, this system is not incidental. Gut microbes shape immune education, barrier function, metabolite availability, inflammatory tone, and even regenerative potential. For that reason, age-related dysbiosis is increasingly difficult to dismiss as a mere downstream correlate of aging. At the same time, it should not be elevated into a universal primary cause. The most defensible placement is as an amplifier layer: a context-sensitive ecological regulator that can intensify, stabilize, or occasionally relieve age-related decline once deeper host vulnerabilities are already present [70].
This placement follows directly from the kind of evidence now available. Reviews of age-related microbial dysbiosis argue that changes in microbial composition and function are mechanistically relevant to mammalian aging rather than epiphenomenal [70]. What makes this plausible is not simply that old organisms harbor different microbes than young ones, but that those microbial shifts plausibly alter host physiology in directions already implicated elsewhere in the aging hierarchy. Dysbiosis can change immune tone, reshape metabolite pools, weaken mucosal barriers, alter nutrient handling, and modulate repair signaling. In other words, the microbiome is well positioned to tune several of the same organism-wide variables already identified as amplifiers—especially inflammation, metabolic stress, and regenerative competence. This is why the microbiome belongs neither in the deepest causal layer nor among passive readouts. It occupies a middle position with real causal leverage.
The strongest reason to avoid treating the microbiome as a mere bystander is that experimental manipulation can improve aging-related function. Fecal microbiota transplantation from young mice rejuvenates aged hematopoietic stem cells, directly linking gut ecology to one of the central regenerative and immune compartments of the body [71]. That result is important because it shows a cell-nonautonomous route by which microbial state can influence stem-cell aging. The effect is not confined to hematopoiesis. Rejuvenating fecal microbiota transfer has also been shown to enhance nerve repair in aged animals, broadening the microbiome’s relevance beyond gut or immune physiology into tissue repair and regeneration more generally [74]. These interventions do not imply that the microbiome sits above all other aging mechanisms. But they do show that changing microbial ecology can shift the functional output of aged tissues in a more youthful direction. That is a hallmark of an amplifier layer.
The microbiome’s importance also becomes clearer when viewed through the lens of inflammaging. Earlier sections argued that chronic low-grade inflammation is a principal route by which many local lesions are translated into organism-wide aging syndrome [48,49]. The microbiome is one of the systems most capable of modulating that inflammatory baseline. Aged host tissues can alter gut permeability, immune surveillance, mucus composition, and epithelial maintenance, thereby reshaping the ecological niche available to microbes. In turn, altered microbial communities can reinforce inflammatory signaling, change innate immune stimulation, and disturb metabolic homeostasis. This is why dysbiosis should be understood as a feedback amplifier rather than a one-way influence. It both responds to host aging and feeds back into it.
That reciprocity is captured especially well in the emerging link between gut dysbiosis and senescence. The recent review on cellular senescence and gut dysbiosis frames these two processes as mutually reinforcing [75]. Senescent cells can alter tissue environments, secretory profiles, and barrier properties in ways that favor dysbiosis, while dysbiotic microbial products and inflammatory tone can in turn promote senescence or stabilize senescence-associated pathology [75]. This is theoretically significant because it connects the microbiome directly to the antagonistic-response and amplifier layers already described. The microbiome is not acting alone. It is participating in a network of chronicity, one in which senescence, inflammation, tissue barrier decline, and microbial ecology lock one another into a more aged state.
The biomarker literature provides a complementary but slightly different kind of support. Microbiome features have been linked to epigenetic age acceleration and physical fitness, suggesting that gut ecology tracks meaningful aspects of biological age rather than merely chronological time [72]. Large-scale gut microbiome data can also be used to construct aging clocks with substantial predictive accuracy [73]. These results matter for two reasons. First, they reinforce that microbial composition and function are not random noise around aging, but structured parts of aging state. Second, they also warn against overclaiming. A strong microbiome clock, like any other clock, does not by itself establish causal primacy. The microbiome may reflect many host processes at once—diet, immune tone, medication exposure, barrier integrity, inflammation, activity, and disease burden. Its value as a predictor therefore supports its relevance, but not necessarily its placement at the root of aging [72,73].
That caution is especially important in a unified theory spanning Animalia. Aging is far older, broader, and more conserved than any single mammalian gut ecology. Animals vary enormously in diet, digestive anatomy, microbial dependence, barrier design, and host–microbe architecture. Yet aging-like processes recur across that diversity. This alone makes it unlikely that the microbiome is the universal originator of aging. Rather, the microbiome seems better understood as an ecological layer whose importance rises particularly in animals—especially mammals—in which host physiology is deeply entwined with dense microbial communities. In those systems, microbial ecology can profoundly shape how deeper host lesions are translated into inflammatory, metabolic, and regenerative outcomes. But it remains a tuning system layered on top of more primary host vulnerabilities, not a substitute for them.
The most useful way to conceptualize this is to say that the microbiome modulates tone. It adjusts inflammatory tone, metabolic tone, barrier tone, and regenerative tone. When host maintenance systems are youthful and robust, the microbiome may contribute to resilience and homeostatic flexibility. When host fidelity has already begun to erode—through immune aging, senescence, mitochondrial dysfunction, or tissue damage—the same host–microbe coupling can become destabilizing. Dysbiosis then amplifies pathology not because microbes create aging from nothing, but because they magnify the consequences of existing host vulnerability. This interpretation fits especially well with the evidence from young-to-old microbiota transfer, where changing the microbial layer improves host function without erasing the deeper lesions that made the host susceptible in the first place [71,74].
In the present hierarchy, then, the microbiome is best placed as a potent but context-dependent amplifier. It is mechanistically relevant to mammalian aging [70]; its manipulation can rejuvenate aged hematopoietic stem cells and improve nerve repair [71,74]; it tracks biological-age measures and supports predictive clocks [72,73]; and it participates in self-reinforcing loops with senescence and inflammation [75]. But it is unlikely to be a universal aging originator across animals, both because aging clearly arises in systems with very different microbial ecologies and because the strongest effects of dysbiosis are best understood as amplification of deeper failures already underway. The microbiome thus fits naturally into the biological-fidelity framework as a host–ecological layer that tunes inflammatory, metabolic, and regenerative tone inside a larger aging architecture. The next section examines another way in which aging becomes locked in physically rather than only biochemically: through tissue mechanics, extracellular matrix drift, and nuclear architecture.
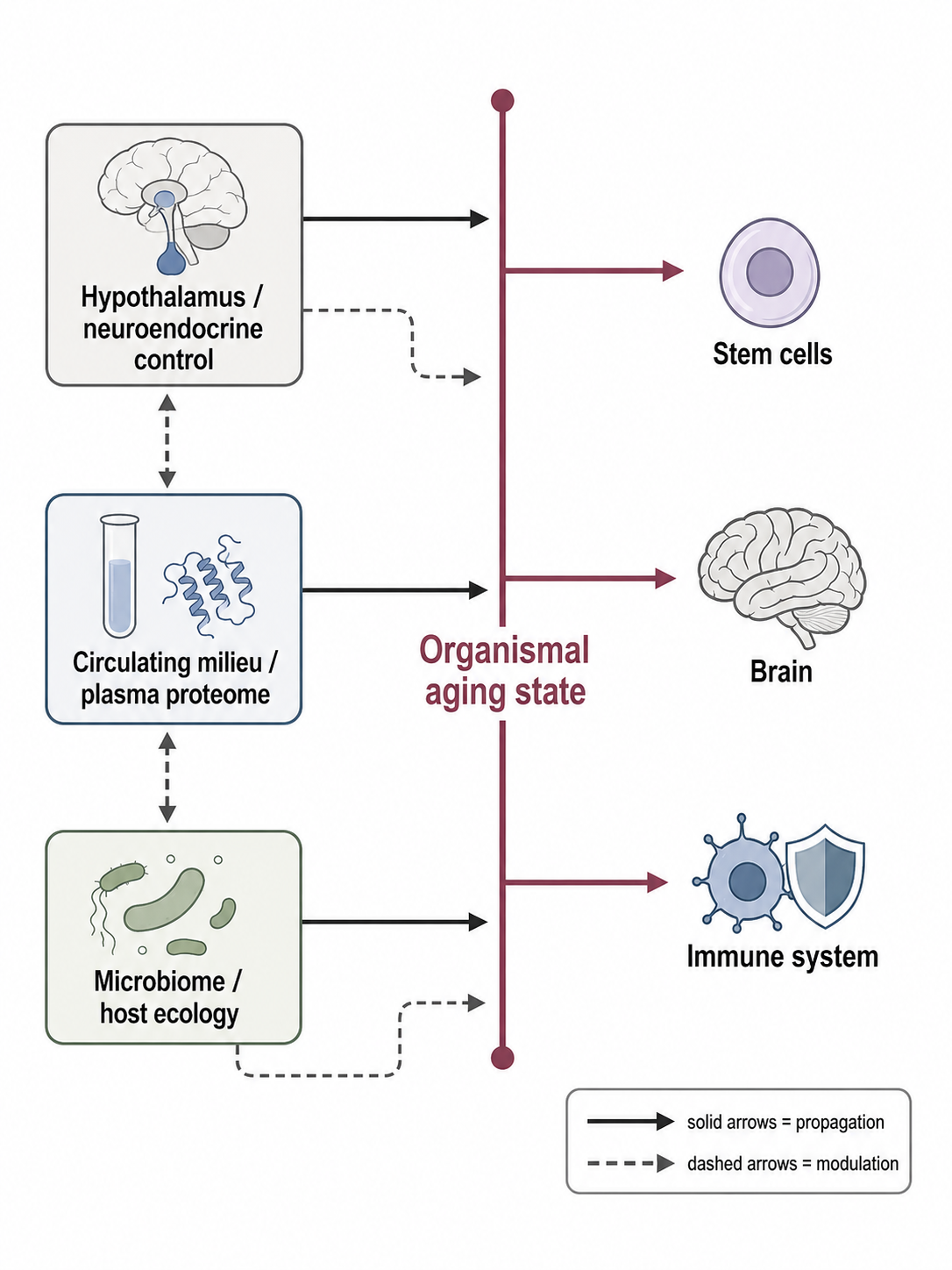
Figure 5. System-wide coordination of aging state. Neuroendocrine control, circulating proteomic milieu, and host–microbe coupling help synchronize local failures across tissues, explaining why aging behaves as a whole-organism state rather than merely a collection of independent local declines.
Amplifier-to-executor bridge: Tissue mechanics, ECM drift, and nuclear architecture
By this stage of the hierarchy, aging is no longer only a problem of damaged molecules, altered signaling, or dysregulated circulating state. It has begun to acquire physical form. This is the distinctive importance of extracellular matrix dynamics, tissue mechanics, and nuclear architecture: they translate diffuse biological aging signals into durable local tissue states. Earlier layers explain how biological fidelity is lost, how maintenance fails, and how inflammatory and systemic signals propagate. The present layer explains how those changes become materially stabilized within tissues. In this sense, extracellular matrix drift and nuclear architectural change form the bridge between amplification and execution. Aging becomes harder to reverse when it is embedded not only in cell state and blood-borne factors, but in the structure and mechanics of the tissue itself [67].
The extracellular matrix is therefore not well described as a passive scaffold that merely records prior damage. It is an active regulatory environment that shapes cell survival, migration, differentiation, inflammatory signaling, and tissue repair. The emerging view that ECM dynamics constitute an underappreciated hallmark of aging is important precisely because it corrects an old bias in the field: age-related fibrosis, stiffening, crosslinking, and compositional change were often treated as downstream scarring, whereas they are increasingly recognized as processes that can actively drive dysfunction [67]. That interpretation is strengthened by comparative biology. The naked mole-rat’s extraordinary accumulation of high-molecular-mass hyaluronan, originally linked to cancer resistance, shows that extracellular matrix chemistry can be part of a species-level longevity strategy rather than merely an inert consequence of life history [81]. This does not mean that matrix composition alone explains aging, but it does mean that tissue architecture belongs inside mechanism, not outside it.
What gives the ECM such leverage is its ability to regulate cell behavior through mechanical state. Cells do not respond only to growth factors and metabolites; they respond to stiffness, tension, confinement, and the viscoelastic properties of their niche. Aging-associated changes in matrix organization and stiffness therefore feed directly into how cells interpret their environment and what transcriptional and epigenetic states they can sustain. A particularly clear demonstration comes from cartilage, where age-related matrix stiffening epigenetically regulates α-Klotho and promotes cartilage aging [68]. This is a crucial result because it shows that mechanics can act upstream of gene regulation. The anti-aging factor α-Klotho is not merely lost because the tissue is old; the aged tissue’s mechanical state participates in repressing it. Matrix stiffening thus becomes more than structural deterioration. It becomes a signaling language through which aging tissues impose a more aged phenotype on their cells [68].
The same logic applies to regenerative niches. In muscle, matrix stiffness drives stem-cell dysfunction through TRAIL signaling, indicating that the aged niche can actively push stem cells away from youthful regenerative behavior [69]. This is especially important for the theory as a whole because stem-cell failure is one of the main executor layers of aging, yet this evidence shows that part of stem-cell aging is not cell-intrinsic in the narrow sense. It is imposed by the material properties of the surrounding tissue. Aging therefore becomes self-maintaining locally: once the matrix has drifted far enough from its youthful state, even stem cells that retain partial intrinsic competence may be unable to execute youthful repair programs [69]. The niche itself has become old, and it teaches age to the cells inside it.
At this point, the nucleus becomes central. The nuclear lamina is the structure through which tissue mechanics and chromatin organization are physically linked. Progeroid laminopathies such as Hutchinson–Gilford progeria syndrome are instructive here because they show what happens when nuclear architecture is severely disturbed: chromatin organization, DNA repair, mechanotransduction, and tissue maintenance fail in ways that generate a compressed aging-like syndrome [87]. The importance of this is not that normal aging is simply a weak laminopathy. It is that the nuclear envelope and lamina are not peripheral supports. They are part of the causal interface between tissue structure and gene regulation. Once that interface is destabilized, aging-like phenotypes can emerge with unusual speed and breadth [87].
The role of the lamina is also visible in normal and senescent tissues. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and chromatin landscape, showing that nuclear architectural decline is sufficient to reorganize regulatory state on a broad scale [112]. In the aging hippocampus, Lamin B1 decline underlies the loss of adult neurogenesis, directly linking age-related nuclear structural change to a specific tissue-level functional deficit [113]. These studies are especially valuable because they connect several themes of the paper at once: epigenetic information loss, cell-state drift, and tissue-specific execution. The nuclear lamina is not only a mechanical buffer; it is a regulator of chromatin accessibility, genome organization, and lineage-relevant transcriptional control. When it declines, cells may no longer be able to preserve the same regulatory architecture even if the underlying DNA sequence remains largely intact [112,113].
This suggests a more general principle: aging tissues become self-maintaining when altered mechanics and altered nuclear architecture begin to lock cells into aged states. Earlier sections argued that senescence, inflammation, and systemic milieu can push cells toward dysfunction. The present layer explains why those pushes may become locally persistent. An aged matrix changes the forces applied to cells; those forces alter cytoskeletal and nuclear organization; altered nuclear organization reshapes chromatin and transcription; the resulting cells further remodel the matrix and tissue environment. In this way, mechanical drift can convert transient or partially reversible states into stable local attractors. The tissue no longer merely contains aged cells. It becomes an aged microenvironment that continually reproduces aging phenotypes.
There is evidence, however, that these states are not absolutely fixed. Aging can induce a hyper-quiescent chromatin state that is reversed by regeneration [116]. That result is important because it implies that aged nuclear and chromatin architecture can, under the right conditions, be remodeled back toward a more youthful organization. In the context of this section, it suggests that tissue mechanics and nuclear structure are not terminal endpoints so much as stabilized states with partial plasticity. This is entirely consistent with a layered theory of aging: once mechanical and chromatin states have drifted, they become harder to reverse than soluble signals, but they may still be reversible if regenerative or reprogramming processes are strong enough to rebuild niche and nuclear organization [116].
A similar implication emerges from the recent literature on mesenchymal drift. A 2025 study reported prevalent mesenchymal drift across aging and disease and showed that this state could be reversed by partial reprogramming [119]. While the exact relationship between mesenchymalization and tissue mechanics is not yet fully resolved, the connection is highly suggestive. Mesenchymal-like states are often associated with altered matrix interaction, migratory behavior, stress adaptation, and extracellular remodeling. It is therefore plausible that chronic inflammatory tone, ECM drift, and nuclear architectural change collectively bias aged cells toward these mechanically and transcriptionally permissive states. The point is not that every aged cell becomes “mesenchymal” in a literal sense, but that tissue aging may involve a broader shift toward states stabilized by altered mechanics and architecture [119].
This framing helps explain why tissue aging often appears locally entrenched even when upstream drivers are systemic. Circulating inflammatory factors, microbiome-derived metabolites, and endocrine changes may be widely distributed, but organs do not all fail identically. One reason is that each tissue embodies those signals through its own architecture. Cartilage stiffens differently from muscle; brain niches differ from vascular niches; immune organs remodel differently from epithelia. Once mechanics and nuclear structure begin to change, the general problem of aging is translated into organ-specific execution logic. The aged tissue then imposes a selective and instructive environment on its resident cells, amplifying some pathways and suppressing others. This is why the present layer is best understood as the bridge from amplifier to executor: it explains how broad biological aging becomes materially localized.
In the hierarchy proposed here, then, tissue mechanics, ECM drift, and nuclear architecture are not primary origins of aging, but they are far too consequential to be treated as late passive fallout. ECM remodeling and stiffening can actively regulate gene expression and stem-cell function [67–69]. Progeroid laminopathies reveal the causal importance of nuclear architecture [87]. Lamin B1 decline shows that structural nuclear change can reorganize chromatin and impair tissue function in both senescent and normally aging settings [112,113]. Regeneration and partial reprogramming demonstrate that some of these states remain at least partly reversible [116,119]. The central inference is that tissue aging becomes self-maintaining when altered mechanics and nuclear architecture lock cells into aged states faster than repair and regenerative programs can restore a youthful niche. That local lock-in sets the stage for the next layer, where aging becomes overtly visible as stem-cell exhaustion and regenerative collapse.
| Process | Propagation role |
|---|---|
| Persistent senescence | Converts damage into chronic SASP signaling and tissue dysfunction. |
| Inflammaging | Translates many local lesions into shared low-grade immune activation. |
| Clonal hematopoiesis | Turns somatic evolution into inflammatory myeloid skew. |
| Systemic milieu | Synchronizes local failures through circulating and neuroendocrine signals. |
| Microbiome dysbiosis | Tunes inflammatory, metabolic, barrier, and regenerative tone. |
| ECM / mechanics | Locks aged states through stiffness, force transmission, and nuclear architecture. |
Table 3. Damage-conversion and amplifier loops. These layers convert upstream fidelity loss into persistent, self-reinforcing tissue and organism-level aging states.
Executor layer I: Stem-cell exhaustion and regenerative collapse
Aging does not become overt tissue dysfunction the moment molecular damage begins. Multicellular organisms can tolerate a great deal of upstream injury, regulatory drift, and environmental stress so long as they retain sufficient capacity to repair, replace, and reconstitute damaged cells. This is why stem-cell exhaustion and regenerative collapse are best placed in the executor layer of the present hierarchy. They are not, in most cases, the first origin of aging. Rather, they are the point at which many upstream failures—genomic, epigenetic, metabolic, inflammatory, microbial, and mechanical—become translated into visible loss of tissue maintenance. In other words, regenerative decline is one of the main ways aging becomes anatomically and clinically real [58,99].
This placement also clarifies a frequent ambiguity in the field. “Stem-cell exhaustion” is often read too literally, as if aging simply depletes stem cells numerically until tissues can no longer regenerate. The literature supports a broader and more mechanistically useful view. Aging can reduce regenerative capacity by altering quiescence, activation thresholds, lineage bias, mitochondrial stress responses, niche interpretation, and systemic responsiveness, even when stem or progenitor cells are still present [58,61,63]. Thus, the relevant executor is not only stem-cell loss, but loss of effective regenerative competence. A tissue becomes old in the strong functional sense when the burden of routine turnover, injury response, and repair exceeds what its resident progenitor systems and niches can still achieve.
The foundational stem-cell literature already framed aging in this way. Reviews on aging, stem cells, and tissue regeneration emphasize that tissue homeostasis depends on lifelong renewal and that aging can therefore be understood as declining regenerative capacity rather than only accumulated inert damage [58]. More recent work decoding aging-dependent regenerative decline across tissues extends that logic across organ systems and suggests that loss of repair competence is coordinated but still strongly tissue-specific [99]. This is important for the present theory because it explains how the same upstream architecture can yield heterogeneous organ aging. Different tissues carry different renewal demands, different niche structures, and different tolerances for loss of progenitor function. Once regenerative reserve falls below tissue-specific demand, decline becomes overt.
The muscle system provides one of the clearest demonstrations that regenerative aging is an executor rather than a pure root cause. In the classic heterochronic parabiosis experiments, exposing aged progenitor cells to a young systemic environment rejuvenated their function, showing that at least part of age-related regenerative decline is imposed by the milieu rather than irreversibly encoded inside the cells themselves [57]. Subsequent work showed that rejuvenation of aged muscle stem cells can restore muscle strength [59], and related studies in human muscle stem cells revealed molecular aging phenotypes that are at least partly reversible [60]. These findings matter because they show that regenerative failure sits downstream of broader organismal context. Old muscle stem cells are often not simply gone; they are constrained, misregulated, or insufficiently activated by aged local and systemic conditions.
Supportive mechanistic work strengthens that interpretation. Age-related muscle stem-cell decline has been linked to loss of heterochromatin caused by S-adenosylmethionine depletion, and metabolic restoration can rejuvenate those cells [27]. Separately, aging-associated hyper-quiescent chromatin states can be reversed by regeneration [116]. Together, these findings suggest that stem-cell aging often reflects altered cell state rather than terminal disappearance. In the language of this paper, regenerative collapse is therefore an execution problem emerging from deeper fidelity loss: upstream genomic, chromatin, and metabolic instability progressively push progenitors into less competent states, and the tissue becomes old when those states can no longer sustain adequate repair.
The hematopoietic system offers a second, equally important model. Hematopoietic stem-cell aging is strongly shaped by intrinsic mitochondrial quality-control checkpoints; a mitochondrial unfolded protein response checkpoint has been shown to regulate hematopoietic stem-cell aging [61]. At the same time, hematopoietic aging is deeply influenced by inflammatory niche conditions and lineage-skewing pressures [63]. This means that regenerative decline in blood and immune tissues is not only a consequence of cell-autonomous damage. It is produced by the interaction of stem-cell state, mitochondrial stress handling, inflammatory environment, and altered niche signals. That interpretation is reinforced by intervention evidence showing that depletion of myeloid-biased hematopoietic stem cells can rejuvenate the aged immune system [62]. Again, the implication is not simple stem-cell loss. It is that aged regenerative compartments often persist in distorted configurations that can, at least partly, be remodeled.
The immune system illustrates especially well why this layer belongs in the executor layer. Earlier sections described inflammaging, clonal selection, and systemic amplification. Here their consequences become tangible: once hematopoietic stem and progenitor output is skewed, immune competence, repair coordination, and barrier defense begin to fail, and age-related dysfunction becomes self-reinforcing [62,63]. Thymic involution provides supportive context for this point, as one of the clearest early organ-level changes underlying immunosenescence [52]. The executor is therefore not a single exhausted stem-cell pool in isolation. It is a broader collapse in the organism’s capacity to renew and recompose key tissues under stress.
The microbiome evidence supports the same logic from another angle. Fecal microbiota transfer from young mice can rejuvenate aged hematopoietic stem cells [71], and rejuvenating microbiota transfer can enhance nerve repair in aged animals [74]. These findings are important because they show that aged regenerative decline can be shifted by changing upstream amplifier environments rather than by replacing the stem cells themselves. In other words, regenerative failure is responsive to ecological and systemic state. That is exactly what one would expect if this layer is downstream of earlier amplifiers and not identical to the primary source of aging.
This executor placement also helps explain why regenerative decline is such a powerful threshold phenomenon. Many upstream lesions can accumulate without immediate macroscopic failure if tissues still possess enough regenerative reserve to compensate. But once reserve drops below demand—because stem-cell activation is blunted, lineage balance is distorted, niches are inflammatory or stiffened, chromatin states are maladaptive, or systemic cues are hostile—the tissue can no longer hide its aging. Muscle weakness, immune decline, impaired wound healing, barrier failure, and poor recovery from injury then become visible not because damage has suddenly appeared, but because compensation has become inadequate [58,59,62,63,71,99].
This view also resolves an apparent paradox in the aging field. Regenerative decline is among the clearest features of aging, yet many studies show partial reversibility. That paradox dissolves if regenerative collapse is treated as an executor rather than an initial cause. Executor layers are exactly where one would expect both high phenotypic importance and partial reversibility: they sit downstream enough to determine organ function, but upstream enough that restoring niche conditions, metabolic support, chromatin state, or systemic environment can still recover part of the lost function [57,59,60,62,71,74,27,116]. Aging tissues therefore often fail not because their regenerative cells are wholly destroyed, but because too many upstream distortions have converged on their ability to operate.
In the framework advanced here, then, stem-cell exhaustion and regenerative collapse are the main route by which deeper aging processes become organ-level dysfunction. They are core executors, not earliest causes. Genome instability, epigenetic information loss, proteostatic and mitochondrial decline, senescence, inflammation, clonal skew, microbiome shifts, and tissue mechanics all push regenerative systems toward reduced competence; once repair demand exceeds that competence, overt aging phenotypes emerge [57,58,61,63,71,99]. Importantly, these executor failures often appear before complete cell loss and can involve hyper-quiescence, lineage misallocation, or niche-dependent suppression rather than disappearance alone [27,62,116]. That point sets up the next section directly: many aging cells are not best described as absent or dead, but as increasingly mis-specified. The next executor layer therefore examines cell identity drift, immune dysfunction, and mesenchymalization as another route by which upstream fidelity loss becomes visible biological aging.
Executor layer II: Cell identity drift, immune dysfunction, and mesenchymalization
Stem-cell exhaustion does not fully explain why aged tissues fail. A second executor layer becomes visible once one asks not only whether regenerative cells remain, but whether the cells that do remain still know what they are. Aging increasingly appears to produce a state in which many cells are not absent or dead, but mis-specified: mature cells drift from their lineage-defining programs, progenitors become differentiation-biased, alternative-lineage features emerge, and pathological plasticity rises. This is why cell identity drift belongs alongside regenerative collapse in the executor layer. It is one of the main ways upstream fidelity loss becomes functional tissue dysfunction. The conceptual bridge is strengthened by the ICE model, in which erosion of the epigenetic landscape during DNA repair advanced aging-like phenotypes and was accompanied by cellular exdifferentiation, and by recent reviews arguing that aging-associated plasticity includes dedifferentiation, biased differentiation, lineage infidelity, and preneoplastic drift rather than simple passive decline alone [21,25].
The immune system provides one of the clearest demonstrations of this process. By analyzing two large human PBMC atlases comprising almost 4 million cells, Connolly and colleagues concluded that immune senescence involves a gradual loss of cellular identity across multiple cell types, reflected in increased expression heterogeneity and inappropriate co-expression of markers. This makes immune aging more than a problem of reduced cell numbers or chronic inflammation; it is also a problem of lineage-definition and state fidelity. The broader Human Cell Aging Transcriptome Atlas extends this logic beyond blood by organizing age-associated transcriptomic alterations across thousands of human samples and more than 50 tissue types, suggesting that identity drift is a cross-tissue feature of human aging rather than a specialty of one organ. At the organ-system level, age-related thymic involution adds a structural explanation for part of this immune dysfunction, because thymic decline reduces thymic cellularity, disrupts the stromal microenvironment, and progressively shrinks the naïve T-cell contribution needed to maintain adaptive immune competence [26,52,98].
The mechanistic importance of higher-order chromatin architecture is especially evident in lymphoid aging. In old bone marrow pro-B cells, large-scale three-dimensional chromatin reorganization distinguishes aged from young cells, with increased compartment-level interactions and reduced interactions within topologically associated domains. The Nature Cell Biology study further linked this reorganization to the key B-cell regulator Ebf1, which shifts from compartment A to B with age, and to weakened interactions across the immunoglobulin heavy-chain locus, changes that impair B-lymphopoiesis. This is a strong example of identity drift as an executor: the cells still exist, but the regulatory architecture that permits faithful B-cell development has been reorganized into a less competent state. A parallel logic appears in aged muscle stem cells, where depletion of intracellular SAM leads to loss of heterochromatin, while restoration of SAM restores heterochromatin content to youthful levels and rejuvenates age-associated features. In both cases, the relevant lesion is not simple cell death, but erosion of the chromatin structures that stabilize correct lineage behavior [28,27].
These observations connect executor biology directly back to the deep epigenetic layer developed earlier in the paper. Aging does not merely alter gene expression noisily; it appears to change the state space available to cells. The review on cellular plasticity during aging emphasizes that age-related shifts can take the form of dedifferentiation, biased differentiation, acquisition of features from alternative lineages, and entry into preneoplastic states, with both cell-intrinsic and cell-extrinsic drivers contributing. Supporting this, work on aging chromatin has shown that a hyper-quiescent chromatin state can form during aging and that regeneration can reverse aspects of this state while rejuvenating molecular and physiological features. That finding is important because it suggests identity drift is stabilized, but not always irreversibly fixed. Aged cells may therefore fail not because their lineage program is erased absolutely, but because it is progressively crowded out by refractory chromatin configurations that make youthful identity harder to maintain and harder to re-enter [25,116].
One of the most provocative attempts to generalize this executor logic across organs is the recent concept of mesenchymal drift. Lu and colleagues analyzed gene-expression data from more than 40 human tissues and 20 diseases and reported a pervasive upregulation of mesenchymal genes across multiple cell types, together with altered stromal-cell composition. Increased mesenchymal drift correlated with disease progression, reduced patient survival, and elevated mortality risk, while suppression of key mesenchymal-drift transcription factors led to epigenetic rejuvenation. Most importantly for this paper’s logic, Yamanaka-factor-induced partial reprogramming markedly reduced mesenchymal drift before dedifferentiation and gain of pluripotency, rejuvenating the aging transcriptome at cellular and tissue levels. This does not prove that mesenchymal drift is the universal final common pathway of aging, but it makes it a serious candidate for one generalized manifestation of cell-state erosion across tissues and diseases [119].
Interpreted cautiously, mesenchymalization in aging need not mean that every aged cell literally converts into a bona fide mesenchymal lineage. The deeper point is that many aged cells may adopt wound-like, fibrotic, migratory, stress-tolerant, or stromalized programs inappropriate for their original physiological role. This fits well with the broader plasticity literature, which argues that aging increases the prevalence of biased differentiation and alternative-lineage features, and with the mesenchymal-drift work showing that stromal and mesenchymal gene modules rise across many tissues and age-related diseases. In this framework, mesenchymal drift is less a narrow cell-biological label than a candidate systems-level signature of identity failure in chronically stressed tissues [25,119].
The next question is why these drifted states persist. Earlier sections already identified two likely stabilizers: chronic inflammation and altered tissue mechanics. The immune-surveillance side of that equation is supported by findings that senescent cells upregulate the T-cell checkpoint molecule PD-L1, helping them evade immune clearance and accumulate in aging tissues. Persistence of senescent or otherwise pathological cell states therefore reflects not only their creation, but failure of their removal. On the mechanical side, age-related matrix stiffening has been shown to drive Klotho promoter methylation, downregulate Klotho, and accelerate chondrocyte senescence, while age-related ECM stiffness in muscle hinders regeneration by inducing intrinsic MuSC dysfunction through TRAIL signaling. Together, these results support the idea that drifted identities are not free-floating transcriptomic oddities. They are stabilized by inflammatory and mechanical niches that continually teach cells the wrong state and make reversion increasingly difficult [56,68,69].
The executor logic of this section is therefore distinct from that of Section 16 but complementary to it. Section 16 argued that tissues fail when regenerative reserve becomes inadequate. Section 17 argues that tissues also fail when the cells that remain become increasingly incorrectly specified. Immune cells lose identity and become noisier and less appropriately differentiated; lymphoid development is distorted by higher-order chromatin changes; tissue cells acquire maladaptive plasticity; mesenchymal-like programs spread across aging and disease; and inflammatory-mechanical environments help lock those states in place. The visible phenotype of aging cells is thus often not “dead” but “mis-specified.” That conclusion sets up the final executor layer naturally: once cells are both less regenerative and less correctly specified, organ-specific failure modules begin to dominate the phenotype of aging in muscle, brain, vasculature, immune organs, cartilage, and barrier tissues [21,25,26,28,98,119].
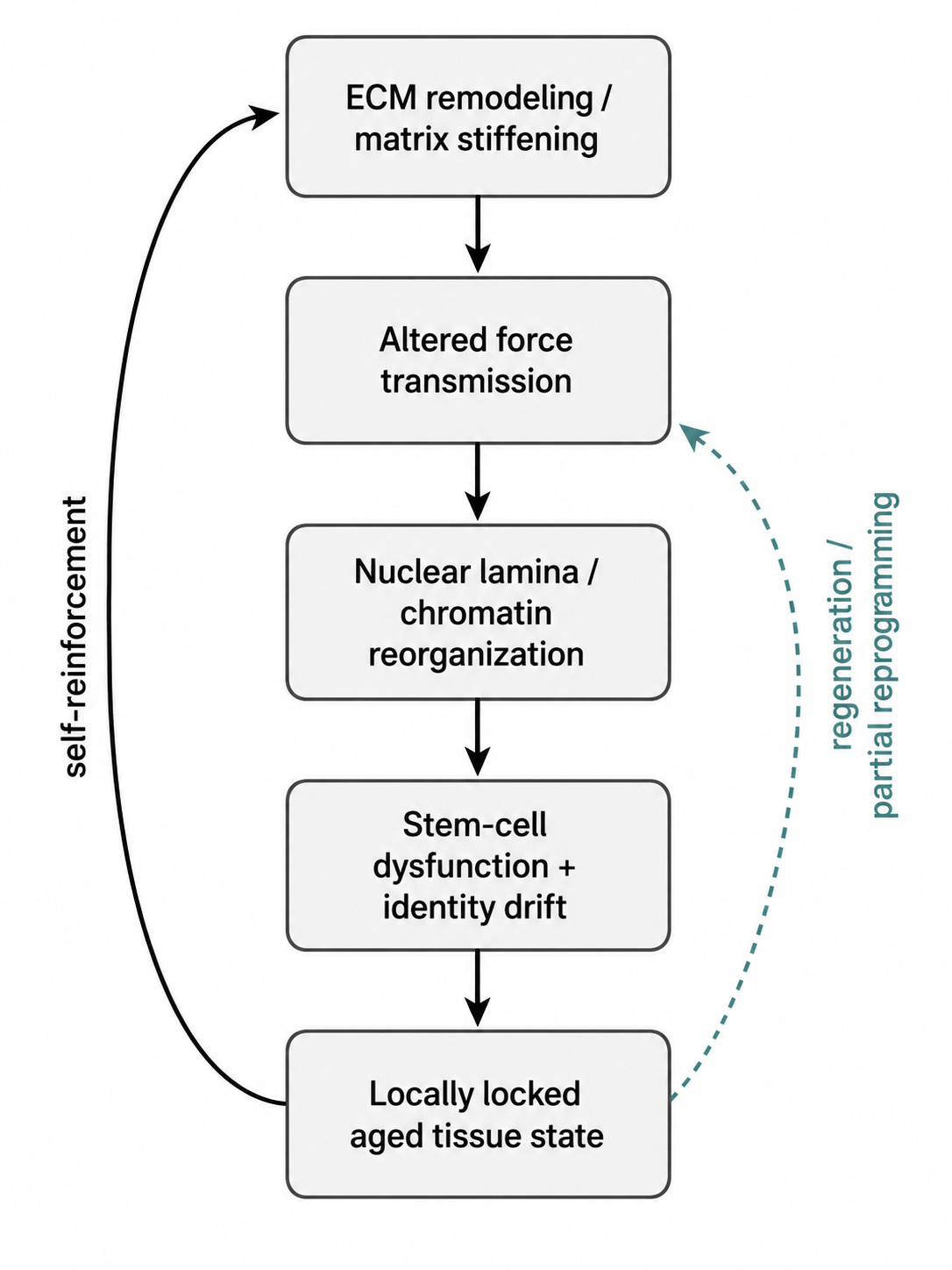
Figure 6. Tissue aging becomes physically stabilized when extracellular-matrix drift and altered mechanics reorganize nuclear architecture and chromatin state, driving stem-cell dysfunction and cell-identity drift that then reinforce the aged niche.
Final executor layer: Organ-specific failure modules
A unified theory of aging cannot stop at shared upstream mechanisms, because organisms do not age as homogeneous masses. Single-cell atlases already show that aging is coordinated yet strongly tissue- and cell-type-specific. Tabula Muris Senis generated lifespan single-cell profiles across 23 mouse tissues and organs, and a companion analysis of that dataset identified aging-dependent genes in 76 tissue-cell types, revealing both shared and tissue-cell-specific aging behaviors [96,97]. The Human Cell Aging Transcriptome Atlas extends this logic into humans by compiling 92 million cells and 3,475 tissue-level samples across more than 50 tissue types, while regenerative mapping across organs shows that regeneration itself varies across tissues and declines with age through distinct local bottlenecks [98,99]. These findings establish the empirical need for a final executor layer: the common aging process must eventually be translated into different organ-specific failure modules.
A reasonable inference from these datasets is that organ-specific phenotypes do not require organ-specific root causes. Rather, common upstream losses of biological fidelity are filtered through tissue architecture: turnover rate, regenerative reserve, stromal geometry, extracellular matrix mechanics, mitochondrial demand, vascular dependence, and exposure to environmental or microbial stress all differ by organ. The same deeper process can therefore culminate in sarcopenia in muscle, involution in immune organs, stiffness-driven degeneration in cartilage, and synaptic or vascular decline in the brain. Organ heterogeneity is thus not evidence against unification; it is the expected anatomical expression of one shared causal process acting under different tissue boundary conditions [96,97,99,114].
Figure 7. Organ-specific failure modules. The same upstream fidelity-loss architecture is executed through different dominant tissue bottlenecks, producing distinct organ phenotypes such as sarcopenia, immune involution, cartilage degeneration, and neurovascular decline.
Skeletal muscle provides one of the clearest examples. Age-associated mitochondrial DNA deletion mutations and enzymatic abnormalities accumulate within skeletal muscle fibers and have been linked to intrafiber atrophy and fiber breakage, directly connecting mitochondrial genomic instability to sarcopenic muscle loss [35]. At the same time, muscle regeneration declines because the stem-cell compartment itself becomes defective: Cosgrove and colleagues showed that aged MuSCs are intrinsically impaired in their capacity to repair myofibers and repopulate the stem-cell reservoir, and human muscle stem-cell studies identified conserved mechanisms of stem-cell aging rather than mere disappearance of satellite cells [59,60]. Aging of the local matrix then worsens this executor module further, because ECM stiffening impairs regeneration by inducing intrinsic MuSC dysfunction through TRAIL-linked signaling [69]. In muscle, then, the final phenotype is produced by convergence between mitochondrial failure, regenerative failure, and mechanobiological niche failure, not by any one lesion alone [35,59,60,69].
Immune organs execute aging through a different dominant module. The thymus is essential for adaptive immunity, yet it undergoes age-related involution that compromises immune responsiveness and sharply reduces regenerative capacity after damage [52]. Recent Nature Immunology work showed that aging generates atypical thymic epithelial states that form dense, thymocyte-poor clusters, act as sinks for trophic epithelial signals, and expand after acute injury, thereby worsening repair of the already involuted thymus. The same study linked these structural epithelial changes to age-related fibroblast programs associated with inflammaging, offering a direct mechanistic bridge from organ involution to systemic immune decline. In this organ, aging becomes clinically visible through collapse of the stromal microenvironment that supports naïve T-cell production and immune renewal [52].
Cartilage and other mechanically dominated tissues reveal another executor logic. In aged cartilage, extracellular matrix stiffening is not just a consequence of aging but an active conversion mechanism: Iijima and colleagues showed that increased matrix stiffness drives Klotho promoter methylation, suppresses α-Klotho expression, and accelerates chondrocyte senescence, whereas exposure to a soft matrix restores a more youthful phenotype and improves cartilage integrity [68]. This is theoretically important because it shows how a low-turnover, highly load-bearing tissue can transform shared upstream aging into a distinctive organ phenotype—loss of chondrocyte integrity, stiffness, and osteoarthritic degeneration—through biophysical stabilization of the aged state. The organ fails through its matrix architecture [68].
The nervous system and neurorepair layers show yet another version of execution. In aged mice, rejuvenating fecal microbiota transfer improved peripheral nerve repair and functional recovery after peripheral nerve injury, indicating that age-related failure in this system can be bottlenecked by neuroimmune and host–microbe interactions rather than only by irreversible neuronal attrition [74]. At a broader systems level, plasma proteomics has shown that accelerated brain and vascular aging predict cognitive decline and Alzheimer’s disease progression, and newer organ-specific clocks linked brain and artery aging to synaptic loss, vascular dysfunction, and glial activation [114,120]. The same organ-specific framework also explicitly models lung and intestine, which is consistent with the idea that barrier-facing systems constitute their own execution class because they age under constant environmental and inflammatory exposure [120]. Here again, the same upstream architecture yields a different output: impaired repair, altered neurovascular signaling, and declining cognitive resilience [74,114,120].
This is why plasma proteomics has become so useful for the executor layer. Lehallier and colleagues first showed that human plasma proteome profiles change across the lifespan in an undulating, age-structured manner [103]. Argentieri and colleagues then developed a general proteomic age clock in 45,441 UK Biobank participants using 2,897 plasma proteins and identified 204 proteins that predicted age accurately; proteomic age acceleration associated with 18 major chronic diseases, multimorbidity, and all-cause mortality across diverse populations [115]. These organism-level clocks are highly informative, but their limitation is also revealing: they capture that aging is underway without necessarily specifying which organ-level executor module is dominating first. That localization requires organ-specific readouts [103,115].
Organ-specific proteomic models begin to provide exactly that localization. Oh and colleagues estimated aging in 11 major organs using plasma proteomics across 5,676 adults and found that nearly 20% of people showed strongly accelerated aging in one organ, whereas 1.7% were multi-organ agers; accelerated organ aging conferred 20–50% higher mortality risk, heart aging strongly predicted heart failure risk, and brain and vascular aging predicted Alzheimer’s disease progression [114]. Wang and colleagues expanded this approach in 43,616 UK Biobank participants and validated ten organ-specific clocks across cohorts from China and the United States, explicitly modeling brain, heart, lung, immune system, artery, intestine, liver, kidney, muscle, and pancreas. In that study, accelerated organ aging predicted disease onset, progression, and mortality beyond conventional clinical and genetic risk factors, with the brain and artery clocks particularly linking synaptic loss, vascular dysfunction, and glial activation to cognitive decline and dementia [120]. Goeminne and colleagues further reported interpretable organ-specific and conventional models trained on chronological age, mortality, and longitudinal proteome data, arguing that diseases can be understood as accelerated aging of particular organismal systems [121]. Organ-specific clocks are therefore best treated not as causes, but as highly useful state estimators of where the executor layer is strongest in a given individual [114,120,121].
The larger implication is that organ heterogeneity does not fracture aging into many unrelated processes. It shows where one shared causal architecture is finally translated into phenotype. Muscle ages through energetic and regenerative collapse; immune organs through involution and stromal failure; cartilage through mechanically stabilized senescence; brain and vasculature through synaptic, glial, and vascular dysfunction; barrier-associated systems through their own repair and exposure constraints. Human multi-omics also shows that aging risk changes nonlinearly across the lifespan rather than rising in a smooth linear fashion, suggesting that the dominant executor modules may shift with age rather than unfold at constant speed [35,52,59,60,68,69,74,96,97,98,99,100,114,120,121]. Organ-specific aging is therefore best understood as the final executor layer of a common upstream process acting on tissues with different structures, tempos, and vulnerabilities.
| Organ module | Dominant executor pattern |
|---|---|
| Skeletal muscle | Mitochondrial burden, MuSC decline, niche stiffness, and sarcopenia. |
| Immune organs | Thymic involution, stromal failure, lineage skew, and immune decline. |
| Cartilage | Matrix stiffening, Klotho loss, chondrocyte senescence, and degeneration. |
| Brain / vasculature | Synaptic loss, glial activation, vascular dysfunction, and impaired repair. |
| Barrier systems | Exposure stress, dysbiosis, inflammatory tone, and weakened repair. |
Table 4. Organ-specific executor modules. Shared upstream aging pressures are translated into different organ phenotypes through tissue-specific bottlenecks.
Stress test I: Exceptional longevity and negligible senescence
A serious theory of aging must explain not only why most animals decline, but also why some species decline extraordinarily slowly or appear to escape ordinary senescent trajectories for long periods. Comparative biologists have long argued that aging research cannot rely only on short-lived laboratory standards, because those species are, almost by definition, poor at resisting aging. The more informative strategy is to study the diversity of lifespan solutions across animals, especially species with unusually long, healthy lives. Cross-species longevity reviews make the same methodological point from another angle: molecular signatures extracted from naturally long-lived species should be compared with within-species lifespan-extension mechanisms, because that comparison can reveal which processes are truly central rather than merely local or species-specific [76,77].
One of the strongest comparative constraints is that longevity often scales with the tempo of underlying biological change. Across mammals, the somatic mutation rate per year shows a strong inverse relationship with lifespan, and DNA methylation rates also negatively scale with maximum lifespan in both blood and skin [9,20]. Bat methylation work pushes that logic further into an exceptionally long-lived mammalian clade: DNA methylation accurately predicts chronological age in bats, and across bat species, greater longevity is associated with a slower rate of age-related DNAm change. Taken together, these findings argue that at least part of exceptional longevity is achieved not by inventing a wholly separate biology, but by slowing the rate at which information-damaging change accumulates [9,20,79].
Bats are especially instructive because they combine unusually long lifespan with traits that, in simpler theories, might be expected to accelerate aging. A recent review highlights eight areas of bat biology relevant to aging, including genome sequencing, telomeres and DNA repair, and immunity and inflammation [78]. The important point is not that bats identify one magical anti-aging pathway. Rather, they repeatedly draw attention to the same broad layers emphasized in the present hierarchy: genome maintenance, immune regulation, and other buffering systems that allow a high-performance organism to tolerate metabolic and ecological stress while keeping the pace of aging unusually slow. Their epigenetic tempo is correspondingly reduced, which fits the idea that long life arises from slower fidelity loss plus stronger systemic buffering, not from exemption to aging itself [78,79].
The naked mole-rat provides a second, mechanistically different route to the same conclusion. Review literature describes it as the longest-lived rodent, with a maximum lifespan exceeding 37 years, and emphasizes its delayed-aging phenotype [80]. One of the clearest specific mechanisms identified in this species is extracellular: naked mole-rat fibroblasts secrete extremely high-molecular-mass hyaluronan, more than five times larger than human or mouse HA, and that unusual matrix chemistry mediates an important component of their cancer resistance [81]. This matters for theory because it shows that exceptional longevity can be supported not only by intracellular repair and slower tempo, but also by reinforced extracellular architecture that suppresses malignant selection and stabilizes tissue integrity. In other words, a long-lived species may fortify both deep maintenance layers and the amplifier-to-executor interface [80,81].
Bowhead whales extend the same lesson into the extreme mammalian range. The bowhead whale is estimated to live for more than 200 years and is likely the longest-lived mammal yet known [82]. Comparative genomic analysis of the bowhead whale identified genes under positive selection and bowhead-specific mutations linked to cancer and aging, and highlighted changes in genes related to cell cycle control, DNA repair, cancer, and aging more broadly [82]. The theoretical implication is straightforward: very slow aging in a gigantic, long-lived mammal does not point to a wholly different category of biology; it points to unusually effective protection of the same vulnerability classes already implicated in ordinary aging—genome maintenance, tumor suppression, and regulation of proliferative fidelity [82].
Taken together, bats, naked mole-rats, and bowhead whales do not suggest a single universal “longevity gene.” They suggest repeated reinforcement of a familiar architecture. Long-lived mammals appear to preserve slower epigenetic tempo, slower mutational tempo, stronger genome and tissue maintenance, and more robust cancer-resistance strategies. That is exactly what a fidelity-loss hierarchy would predict: the species that age slowly should be those that either reduce the rate at which fidelity is lost or increase the buffering capacity that prevents local failures from becoming systemic decline [9,20,76–82]. Exceptional longevity therefore functions as a stress test that the theory passes, because the species at the far right tail of lifespan distribution still cluster around the same deep causal layers rather than demanding a completely separate explanatory framework.
Hydra is even more informative because it tests whether aging-like decline can be suppressed to the point of negligible senescence in a simple animal body plan. Hydra reviews emphasize that the genus is unusually valuable precisely because it contains both non-aging and aging-like states: in Hydra oligactis, cold-dependent sexual differentiation can trigger rapid aging and death, whereas other Hydra contexts exhibit continued self-renewal and long-term maintenance [83]. A 2015 study reported constant mortality and fertility over age in Hydra, directly challenging the claim that increasing mortality and declining reproduction are inevitable after maturity [84]. Yet Hydra does not merely tell us that “some animals do not age.” More importantly, it tells us which biological layers are crucial when aging-like decline does appear. In aging Hydra oligactis, lack of epithelial stem-cell renewal and deficient epithelial autophagy were identified as major causes of decline [85]. So even negligible senescence does not fall outside the hierarchy; it points to a condition in which renewal and maintenance systems remain sufficiently intact that fidelity loss fails to avalanche into organismal deterioration [83–85].
The African turquoise killifish provides the opposite extreme and is therefore an essential counterpoint. Its genome paper explicitly frames it as a naturally short-lived vertebrate, and the corresponding full-text snippet notes that, in the laboratory, diapause can be skipped and the fish has a captive lifespan of about 4–6 months, making it the shortest-lived vertebrate model system bred in captivity [111]. This compressed lifespan does not argue against a unified theory. It argues for one. The killifish does not invent a new kind of aging; it compresses vertebrate aging into an experimentally convenient timescale. As a result, it serves as the fast-aging mirror image of bowhead whales and naked mole-rats: a system in which the same broad layers of maintenance, regulation, and execution can be overloaded quickly enough to observe their interactions across an entire vertebrate life course [111].
The comparative conclusion is therefore not that aging is too diverse for unification. It is that diversity reveals the control points of a unified process. Long-lived mammals indicate that slower tempo, stronger repair, and reinforced cancer-resistance and tissue-stabilizing systems can markedly delay aging. Hydra indicates that sustained renewal and autophagy can suppress aging-like decline so strongly that negligible senescence becomes plausible. Killifish shows the converse: when lifespan is compressed, the same architecture runs fast rather than disappearing. Exceptional species therefore do not invalidate a fidelity-loss hierarchy. They identify which layers are most suppressible, most bufferable, and most decisive for determining whether local biological errors remain contained or culminate in organism-wide aging [76–85,111].
| System | Layer implication |
|---|---|
| Bats | Slow epigenetic tempo and immune buffering support delayed fidelity loss. |
| Naked mole-rat | ECM reinforcement and cancer resistance show tissue-architecture buffering. |
| Bowhead whale | Repair and tumor buffering show strengthened maintenance and surveillance. |
| Hydra | Renewal and autophagy can suppress aging-like decline. |
| Turquoise killifish | Compressed lifespan shows the same vertebrate architecture running fast. |
Table 5. Comparative stress tests from exceptional species. Long-lived, negligibly senescent, and compressed-lifespan species identify which layers of the aging hierarchy can be slowed, buffered, or accelerated.
Stress test II: Accelerated-aging states as mechanistic perturbations
Accelerated-aging states are most informative when they are treated neither as literal miniature versions of ordinary aging nor as irrelevant disease-specific curiosities. Their real value lies in their selectivity. They compress time, exaggerate particular lesions, and reveal which parts of the aging architecture can be driven hardest by specific perturbations. Read in that way, they function as natural experiments. If the hierarchical model proposed here is correct, then fast-aging conditions should not generate wholly alien biology. They should enter the system at different points, overload different layers, and nevertheless converge on recognizably age-like patterns of dysfunction. That is, broadly, what the evidence shows [86–95].
The clearest example is Hutchinson–Gilford progeria syndrome (HGPS), which strongly loads the genome–nuclear-architecture axis. Mutant LMNA and the production of progerin disrupt the nuclear lamina, deform nuclear shape, perturb chromatin organization, impair genome maintenance, and accelerate cellular senescence, producing a striking syndrome of early vascular and mesenchymal degeneration [86,87]. The importance of HGPS is not that normal aging can be reduced to “progerin pathology.” HGPS is segmental: it amplifies some age-like phenotypes far more than others, arises from a very specific initiating lesion, and does not recapitulate the full spectrum of ordinary aging [86,90]. But that selectivity is exactly what makes it useful. When a primary disturbance at the nuclear-envelope/chromatin interface rapidly yields DNA-damage responses, senescence, tissue dysfunction, and systemic fragility, it argues strongly that nuclear architecture is not a peripheral feature of aging biology. It is one of the deep structural points at which biological fidelity can fail.
Werner syndrome complements rather than duplicates HGPS. Here the initiating defect lies less in lamina architecture than in DNA metabolism itself. Loss of WRN function compromises replication, repair, recombination, and telomere maintenance, and the resulting phenotype includes cataracts, diabetes, atherosclerosis, osteoporosis, and malignancy—features that in some respects resemble a more adult-onset and clinically distributed progeroid state [88,89]. Werner syndrome therefore places especially heavy weight on genome instability and repair burden. Together, HGPS and Werner syndrome show that progeroid disease is not one mechanism but a family of perturbations concentrated in the deep causal zone where genome integrity, chromatin organization, telomere maintenance, and nuclear structure intersect. The broader review literature on premature-aging disease makes the same point in methodological terms: these disorders are illuminating not because they are perfect replicas of aging, but because they reveal what follows when specific maintenance layers are catastrophically weakened [90].
Their relevance to normal aging is strengthened by the fact that related nuclear-lamina and chromatin changes are not confined to rare monogenic disease. Lamin B1 depletion in senescent cells induces large-scale shifts in gene expression and chromatin landscape, and age-associated lamin decline contributes to impaired adult hippocampal neurogenesis [112,113]. This does not collapse physiological aging into LMNA syndrome. It does, however, show that the bridge from progeroid nuclear pathology to normal aging is biologically real. Severe lamina disruption accelerates breakdown of cell-state stability; milder age-linked lamina erosion appears to participate in the slower version of the same logic [87,112,113]. In the language of this paper, progeroid syndromes strongly load the deep genome/nuclear-architecture layer and thereby expose how failures in that layer can cascade outward.
Cancer therapy provides a different sort of accelerated-aging model and one that is clinically widespread rather than genetically rare. Here the initiating insult is acute, imposed, and overtly damaging: radiation and cytotoxic treatment create a large burden of DNA damage, tissue injury, niche disruption, and regenerative stress. Yet the syndrome that follows often persists long after therapy ends. Survivors show frailty, diminished physiological reserve, cardiovascular and metabolic disease, impaired tissue repair, immune dysfunction, and broader multimorbidity commonly associated with later life [91]. The crucial mechanistic point is that cancer therapy does not primarily overload the deepest maintenance layer in the same way as HGPS or Werner syndrome. It overloads the conversion layers: senescence, tissue damage, and chronic aftereffects of the damage response. Persistent therapy-induced senescent cells are particularly important in this regard, because they turn a transient exposure into a durable pathological state [92].
This makes medical acceleration models especially revealing for causal ranking. They do not show that senescence is the primordial source of aging. Rather, they show how powerful senescence is as a converter of insult into organismal decline. A pulse of damage can become a long-lived aging-like syndrome if senescent cells persist, stem-cell compartments are blunted, and damaged tissues fail to reset to a pre-injury state [91,92]. The broader literature on innate danger sensing helps explain how this persistence can become systemic. DNA damage and chromatin disruption can activate cGAS–STING signaling, while retrotransposon derepression can reinforce inflammatory loops that outlast the initiating lesion [50,55]. In other words, cancer therapy is a particularly strong argument for the middle layers of the hierarchy: the antagonistic response and amplifier systems are fully capable of pushing tissues toward an aged state once they are chronically engaged.
Obesity and chronic nutrient overload test yet another entry point into the hierarchy. They do not begin with a rare mutation in DNA repair or nuclear-envelope proteins, nor with an acute iatrogenic burst of tissue damage. Instead, they chronically distort nutrient sensing, adipose signaling, inflammatory tone, mitochondrial function, extracellular-matrix remodeling, and tissue repair. Reviews linking obesity to hallmark biology and the concept of “adipaging” show broad overlap with age-associated processes: chronic low-grade inflammation, senescence-like changes in adipose and stromal compartments, altered metabolic regulation, stem/progenitor dysfunction, fibrosis, and increased burden of age-related disease [93,94]. The theoretical importance of obesity is therefore directional. It suggests that aging can be accelerated not only from below, by destabilizing core maintenance systems, but also from above, by chronically overdriving the regulatory and systemic layers that determine how maintenance is allocated and how local stress is propagated.
Obesity is not equivalent to aging, and its effects are heterogeneous, context-dependent, and at least partly reversible. But that limitation is instructive rather than disqualifying. If aging were nothing more than passive, unavoidable molecular wear, chronic nutrient excess should not so effectively reproduce age-like inflammatory, regenerative, and structural deterioration. Instead, obesity supports the claim advanced earlier in this paper: conserved allocation systems and amplifier loops can materially alter the pace and phenotype of aging by shifting how quickly biological fidelity loss is converted into tissue dysfunction [93,94]. It is therefore best interpreted as a natural model of accelerated loading of the rate-control and amplifier layers, not as a complete replacement for the deeper causal layers.
Treated HIV infection offers a parallel but immunologically distinct acceleration model. Here the dominant burden falls on chronic immune activation, impaired surveillance, inflammatory persistence, and molecular readouts of accelerated biological aging. Even under effective antiretroviral control, treated HIV is repeatedly associated with immunosenescence-like changes, multimorbidity, chronic inflammatory signaling, and epigenetic-age acceleration [95]. That profile makes HIV especially informative for the amplifier side of the hierarchy. It shows that an organism can be pushed toward an older biological state by sustained disruption of immune regulation and systemic inflammatory tone, even without a classical progeroid mutation and even without the massive acute insult of cancer therapy. The route is different, but the convergence is familiar: higher inflammatory burden, weaker repair ecology, and earlier emergence of age-associated pathology [95].
Taken together, these conditions do not point to separate kinds of aging. They point to different entry points into a shared architecture. HGPS and Werner syndrome disproportionately stress the deep genome/nuclear-architecture layer [86–90]. Cancer therapy disproportionately stresses the senescence-and-tissue-damage conversion layer [91,92]. Obesity disproportionately stresses nutrient-sensing, metabolic, and inflammatory amplification [93,94]. HIV disproportionately stresses chronic immune dysregulation and biological-age acceleration [95]. No single model reproduces the whole phenotype of ordinary aging because no single model loads all layers equally. But that pattern is exactly what a hierarchical theory predicts. Normal aging is not one lesion marching linearly outward. It is a multilevel process whose manifestation depends on where fidelity is first destabilized, how strongly protective responses are engaged, and how effectively amplifier loops propagate local disturbance into organism-wide state change.
The segmental nature of accelerated-aging states is therefore not a problem for unification. It is evidence for it. If these conditions produced wholly unrelated outcomes, or if each required an entirely separate conceptual framework, then the case for a unified theory would weaken. Instead, they show organized diversity. Nuclear-envelope and DNA-maintenance disorders accelerate chromatin and repair failure [86–90]. Iatrogenic damage accelerates senescence, niche injury, and chronic inflammatory aftereffects [91,92]. Metabolic overload accelerates nutrient-sensing dysregulation and inflammaging-like systemic stress [93,94]. Chronic infection accelerates immune aging and molecular age readouts [95]. The perturbations differ, but the routes of propagation converge.
This leads to the key inference. Accelerated-aging states map onto the normal aging hierarchy rather than inventing separate biology. They are best understood as mechanistic perturbations that overload particular layers of a common system. That conclusion matters because it distinguishes core mechanisms from passengers. Progeroid syndromes indicate that genome maintenance and nuclear architecture sit unusually deep in the causal stack. Cancer therapy indicates that senescence and damage persistence are powerful converters of insult into aging-like decline. Obesity and HIV indicate that systemic metabolic and immune dysregulation can materially accelerate aging from the regulatory and amplifier levels. In short, these stress tests do not undermine a unified theory. They refine it, by showing that the same organism-wide syndrome can be reached through different but intelligible paths within one multiscale architecture of fidelity loss [86–95].
| Perturbation | Layer overloaded |
|---|---|
| HGPS / Werner | Deep genome-maintenance and nuclear-architecture layers. |
| Cancer therapy | Damage persistence, senescence, SASP, and tissue injury. |
| Obesity | Metabolic rate-control and inflammatory amplifier layers. |
| HIV | Immune activation, immunosenescence, and clock acceleration. |
Table 6. Accelerated-aging perturbations mapped to the hierarchy. Fast-aging states overload recognizable layers of normal aging biology rather than creating wholly separate mechanisms.
Stress test III: Rejuvenation and partial reversibility
If accelerated-aging states show where the aging system can be driven into premature failure, rejuvenation studies show something equally important: which parts of that failure remain pliable once age-associated decline has already appeared. In theoretical terms, this is a decisive stress test. A model of aging built entirely out of irreversible wear would predict that meaningful restoration should require replacement of cells, organs, or whole tissues, because the central substrate of decline would already be physically spent. But that is not what the current evidence shows. Across reprogramming, regeneration, systemic milieu transfer, metabolic rescue, telomere restoration, and microbiome manipulation, age-associated phenotypes can move in a younger direction without wholesale cell replacement [16,17,22–24,27,39,57,59,71,73,74,104,116,119]. These findings do not imply that all aging is reversible. They do imply that a substantial part of what manifests as aging is encoded in biological states that remain experimentally modifiable.
The strongest evidence comes from partial reprogramming. The retinal-vision study showed that partial reprogramming could recover youthful epigenetic information and restore function in a mammalian system, directly challenging the assumption that age-related loss of cellular state is strictly one-way [22]. A later gene-therapy-based study extended this logic from local rescue toward organism-level outcome by reporting lifespan extension in old mice [23]. Read conservatively, these experiments still do not prove that all age-linked damage has been erased, nor do they justify the crude conclusion that aging is simply a reversible epigenetic switch [24]. Yet even the conservative reading is theoretically powerful: aged cells can be pushed toward a younger functional state without returning them to pluripotency and without replacing them wholesale. That fact is difficult to reconcile with any account in which the dominant substance of aging is nothing but accumulated irreparable wear.
This point becomes sharper when rejuvenation is interpreted through the problem of cell identity. Review work on reprogramming and cellular plasticity increasingly frames aging not just as damage accumulation but as progressive loosening of the regulatory constraints that stabilize differentiated cell state [24,25]. Recent evidence that prevalent mesenchymal drift in aging and disease can be reversed by partial reprogramming fits exactly into that picture [119]. Here rejuvenation is not best imagined as repairing a broken component in isolation. It is better understood as re-imposing the informational rules that keep cells specified, coordinated, and resistant to pathological drift. In the framework advanced in this paper, that matters enormously. It suggests that one of the most reversible fractions of aging lies in the control architecture of identity itself.
Independent evidence from chromatin biology points in the same direction. A hyper-quiescent chromatin state formed during aging can be reversed by regeneration, indicating that at least some age-associated chromatin locking is maintained rather than terminal [116]. In aged muscle stem cells, loss of heterochromatin driven by SAM depletion contributes to functional decline, and metabolic restoration rejuvenates those cells [27]. These results are important because they localize reversibility to the epigenetic-control layer. The chromatin state of an aged cell is not merely a fossilized record of prior insult. At least in part, it is an active configuration that can be remodeled, reopened, and functionally reset [27,116]. That is exactly what one would expect if aging centrally involves erosion of biological information rather than only passive lesion load.
Systemic rejuvenation experiments broaden the same inference from cells to whole organisms. Exposure to a young systemic environment rejuvenates aged progenitor cells [57], and young blood can reverse age-related impairments in cognition and synaptic plasticity [104]. Related work shows that rejuvenation of aged muscle stem cells can restore muscle strength [59]. These interventions do not erase a lifetime of somatic mutation, reconstruct every damaged extracellular matrix, or eliminate all accumulated molecular lesions. Instead, they change the environment in which cells interpret stress, allocate repair, maintain lineage state, and execute regeneration. The existence of strongly age-structured plasma proteome trajectories across the lifespan further supports the idea that systemic age is carried in a coordinated circulating state rather than only in cell-intrinsic damage burden [103]. Rejuvenation from the level of blood and milieu therefore argues that aging is partly a network phenomenon: tissues are old not only because they are damaged, but because they are embedded in an aged signaling environment.
Microbiome interventions extend that logic into host–microbe coupling. Fecal microbiota transfer from young mice can rejuvenate aged hematopoietic stem cells [71], and rejuvenating microbiota transfer can improve nerve repair in aged animals [74]. Large-scale microbiome-based aging clocks indicate that microbial community structure itself carries strong age-related information [73]. The microbiome is unlikely to be a universal origin point of aging, but these results show that it belongs to the reversible amplifier layer. Altering microbial ecology can shift inflammatory tone, regenerative competence, and system-level age phenotype without directly repairing every deeper lesion [71,73,74]. That is exactly what the hierarchy proposed in this paper would predict for an amplifier system: it is not the deepest cause, but it is plastic enough to move function appreciably.
Layer-specific rescue experiments provide the same lesson from a different angle. Telomerase reactivation in telomerase-deficient aged mice can reverse tissue degeneration [16], and telomerase gene therapy in adult and old mice delayed aging and extended longevity in that experimental setting [17]. Likewise, NAD+ repletion improves mitochondrial and stem-cell function and extends lifespan in mice [39]. These interventions are not universal demonstrations that any aged tissue can be restored without limit. Telomerase rescue is especially informative in proliferative and telomere-compromised contexts, while NAD restoration acts more as a reset of metabolic and mito-nuclear coordination than as a literal erasure of all prior damage [16,17,39]. But precisely because these rescues are layer-specific, they are mechanistically clarifying. They show that some aging phenotypes arise from maintenance modules that are not fully exhausted and can still be pushed back toward youthful function.
Taken together, the rejuvenation literature implies that the most reversible fraction of aging lies in regulatory state, intercellular coordination, and maintenance control. Chromatin configuration, lineage identity, stem-cell niche signals, inflammatory tone, mitochondrial communication, and aspects of telomere-linked regenerative competence can all move toward a younger state on experimentally relevant timescales [16,17,22–25,27,39,57,59,71,73,74,104,116,119]. This does not mean that deeper lesions are unimportant. It means that much of their physiological impact is mediated through middle layers that remain plastic. In the language of this theory, irreversible insults accumulate, but they generate a large superstructure of reversible dysregulation.
That distinction is crucial, because rejuvenation findings argue against two simplistic extremes at once. They argue against a purely irreversible wear model, since substantial restoration occurs without wholesale replacement [22,23,27,39,57,71,74,104,116,119]. But they also argue against a purely programmed model, because the reversibility is partial, context-dependent, and clearly constrained. No cited intervention demonstrates complete erasure of somatic mutation burden, universal correction of clonal mosaicism, full reconstruction of heavily remodeled extracellular matrix, or guaranteed restoration of all lost tissue architecture [16,17,22–24,39,57,71,74]. The right conclusion is therefore not that aging is unreal or easily switched off. The right conclusion is that aging contains both a less reversible substrate and a more reversible superstructure.
This layered interpretation also explains why rejuvenation should succeed best before structural damage has crossed critical thresholds. When tissue architecture, cell number, and regenerative niches are still salvageable, resetting chromatin state or systemic signaling can yield large functional gains. When fibrosis, scarring, cell loss, clonal dominance, or mechanical lock-in have become dominant, the same interventions may still help, but they will encounter harder limits. In other words, rejuvenation works when restoration of biological fidelity outruns the degree of irreversible structural deterioration. It becomes weaker as damage is translated into more stable physical reorganization of tissue. This is exactly the pattern one would expect if aging unfolds from deep lesions through plastic amplifier layers into increasingly entrenched executor states.
The regeneration evidence is especially important here, because it links reversibility not only to external intervention but to endogenous restorative programs. If regeneration can reverse a hyper-quiescent chromatin state [116], then the organism itself retains mechanisms capable of re-opening aged regulatory configurations under the right conditions. Rejuvenation is therefore not wholly foreign to biology. It may be an extension or reactivation of repair logics that persist but become insufficient, badly coordinated, or chronically suppressed with age. This fits neatly with the broader theme of the paper: aging is not merely the accumulation of insults, but the progressive failure of fidelity-preserving systems to keep pace with them.
For the unified theory, the main lesson is direct. Rejuvenation works not because all damage can be erased, and not because aging is only a top-down program, but because much of age-related dysfunction is mediated through plastic control layers that sit between deep lesions and terminal tissue failure. Partial reprogramming, young systemic milieu, stem-cell niche rescue, telomerase reactivation, NAD restoration, regeneration-associated chromatin resetting, and microbiome transfer all point to the same conclusion: the reversible share of aging lies most strongly in epigenetic/regulatory state and the systemic amplifier layers, while the less reversible share lies in accumulated structural damage and long-entrenched tissue remodeling [16,17,22–25,27,39,57,59,71,73,74,104,116,119]. That is one of the strongest arguments available against a purely irreversible wear model. Aging is real, damage is real, but the phenotype of aging is not exhausted by damage alone. A large portion of it remains a problem of information, coordination, and state—and that portion can, at least partly, be restored.
| Lever | Predicted earliest movement |
|---|---|
| Partial reprogramming | Epigenetic state, cell identity, and functional plasticity. |
| Telomerase | Renewal capacity in telomere-limited tissues. |
| NAD restoration | Mitochondrial coupling, stem-cell function, and metabolic resilience. |
| Young systemic milieu | Niche signaling, regeneration, cognition, and plasma proteomic state. |
| Microbiota transfer | Microbial age, inflammatory tone, and repair capacity. |
| Senolytics / senomorphics | Late-life senescent-cell burden, SASP, and frailty measures. |
| Rapamycin / CR-like interventions | Rate-control tempo, autophagy, and maintenance investment. |
Table 7. Rejuvenation levers and expected earliest shifts. The theory predicts that interventions should first move the layers they directly target, with regulatory and systemic states often improving before irreversible structural lesions.
Biomarker and readout layer: What clocks see and what they do not explain
Biological-age clocks are one of the most important advances in aging research, and also one of the easiest places for the field to make a category error. Once a molecular pattern predicts chronological age, mortality, disease risk, or intervention response, there is a strong temptation to treat that pattern as the mechanism of aging itself. That temptation should be resisted. What clocks see is real: their success shows that aging is structured, conserved, and measurable across multiple biological layers. But what they see is not automatically an explanation. A clock can be highly predictive and still fail to identify the deepest cause, the causal order among mechanisms, or the distinction between upstream drivers, compensatory responses, amplifier loops, and downstream executors [4]. In the framework advanced here, clocks belong to the readout layer. They matter precisely because aging is not random noise. Yet a state estimator is not the same thing as the generator of the state it estimates.
DNA methylation clocks provided the clearest early demonstration that biological age is a real and organized property of living systems. Horvath’s pan-tissue clock showed that DNA methylation patterns track age across many human tissues and cell types [18]. This was not merely a technical achievement. It implied that age-linked biological change leaves a reproducible signature across cellular contexts that differ enormously in function, turnover, and environment. Later comparative work deepened that conclusion. A mammalian methylation clock showed that age-linked methylation structure is conserved across mammals [19], and subsequent analysis showed that methylation rates themselves scale with maximum lifespan across mammalian species [20]. Together, these findings strongly argue that aging is associated with a conserved epigenetic tempo rather than a purely local or species-specific collection of accidents [18–20]. If aging were nothing but uncoordinated molecular decay, the existence of such stable, cross-tissue, and cross-species methylation regularities would be hard to explain.
Outcome-oriented methylation measures pushed this even further. GrimAge strongly predicts lifespan and healthspan [109], while DunedinPACE was designed to estimate the pace of aging rather than only the accumulated biological state associated with age [110]. This distinction is conceptually important. Some biomarkers mostly summarize how far along an age trajectory a system appears to be. Others attempt to capture how fast that trajectory is currently unfolding. In a causal hierarchy, those are not interchangeable measurements. Rate-control pathways should be expected to affect pace measures earlier or more strongly than they affect markers of cumulative burden, whereas irreversible structural damage may continue to limit function even after pace has shifted. The existence of both kinds of clocks is therefore useful because it gives the field a way to separate age state from aging rate [109,110]. But it still does not follow that the clocks themselves are the mechanisms. A variable can be an excellent summary of a process without being the cause of the process it summarizes [4].
The proteomic literature extends the same logic into the systemic and organ-specific domain. Plasma proteome profiles change in structured, nonlinear ways across the lifespan [103]. More recent work has shown that plasma proteins can report organ-aging signatures, track aging and disease, predict mortality and risk of common age-related diseases across diverse populations, and generate organ-specific biological aging models [114,115,120,121]. These studies are especially important for a systems theory of aging because they show that biological age is not merely an abstract scalar. It can be decomposed, at least partly, into anatomically meaningful system states. Different organs can appear biologically older or younger than expected, and disease can be interpreted as accelerated aging of particular organismal systems [114,120,121]. This fits well with the broader framework of the paper, in which shared upstream processes are expressed through tissue-specific executor modules. But once again, predictive power does not dissolve the distinction between readout and cause. The plasma proteome integrates inflammatory signaling, endocrine state, tissue damage, repair responses, cell-composition shifts, metabolic status, and organ cross-talk. Its very strength as a biomarker comes from integrating many layers at once. That makes it an excellent state estimator, but also ensures that it is not a simple causal primitive [103,114,115,120,121].
Microbiome-based aging measures reinforce the same lesson from a different part of the system. Microbiome features have been linked to epigenetic age acceleration and physical fitness [72], and large-scale gut microbiome data can be used to build accurate aging clocks [73]. This shows that age-associated biological structure extends beyond host cells into host–microbe ecology. In other words, organismal aging is reflected not only in methylation patterns and plasma proteins, but also in the composition and organization of microbial communities coupled to the host [72,73]. That is theoretically important because it supports the view that aging is a whole-system state distributed across interacting compartments. At the same time, it illustrates why clocks cannot be equated with origins. A microbiome clock may reflect immune aging, barrier dysfunction, altered metabolism, ecological feedbacks, or lifestyle context. It captures biological age at one amplifier layer; it does not, by itself, reveal whether the microbiome is an initiator, a propagator, or mostly a reporter in any given setting [72,73].
Single-cell and multi-omic atlases help explain why clocks can be so informative while remaining mechanistically incomplete. Tabula Muris Senis and the Human Cell Aging Transcriptome Atlas show that aging is coordinated across the organism yet strongly cell-type-specific and tissue-specific in its local expression [96,98]. Multi-omic analyses further suggest that human aging trajectories are not simply linear drifts but may include nonlinear transitions or phase-like reorganizations [100], a picture that is compatible with the undulating changes seen in the plasma proteome across the lifespan [103]. These findings imply that biological age is not a single substance hidden in one molecular layer. It is better understood as a compressed description of position within a multiscale state space generated by many coupled processes. Some changes are broadly shared. Others are tissue-restricted. Some emerge gradually; others appear in coordinated inflection-like shifts [96,98,100,103]. Clocks work because they project this high-dimensional structure onto lower-dimensional measurements. They are successful because the underlying biology has order. But projection and explanation are not the same thing.
This is exactly why the warning from the critical clocks review remains so important: clocks are powerful measurements, but they are not automatically mechanistic explanations [4]. The very properties that make a clock useful often make it causally ambiguous. A methylation clock may be especially sensitive to epigenetic-state drift, but it may also capture downstream changes in cell composition, inflammatory exposure, or altered lineage stability. A proteomic clock may reflect organ stress, systemic inflammation, endocrine coordination, senescence burden, or regenerative decline. A microbiome clock may reflect both host physiology and ecological feedback [4,72,73,114,121]. None of those possibilities invalidates the biomarker. On the contrary, a composite signal is often exactly what makes the biomarker robust. But it does mean that no clock, simply by virtue of prediction, can rank the causal hierarchy on its own.
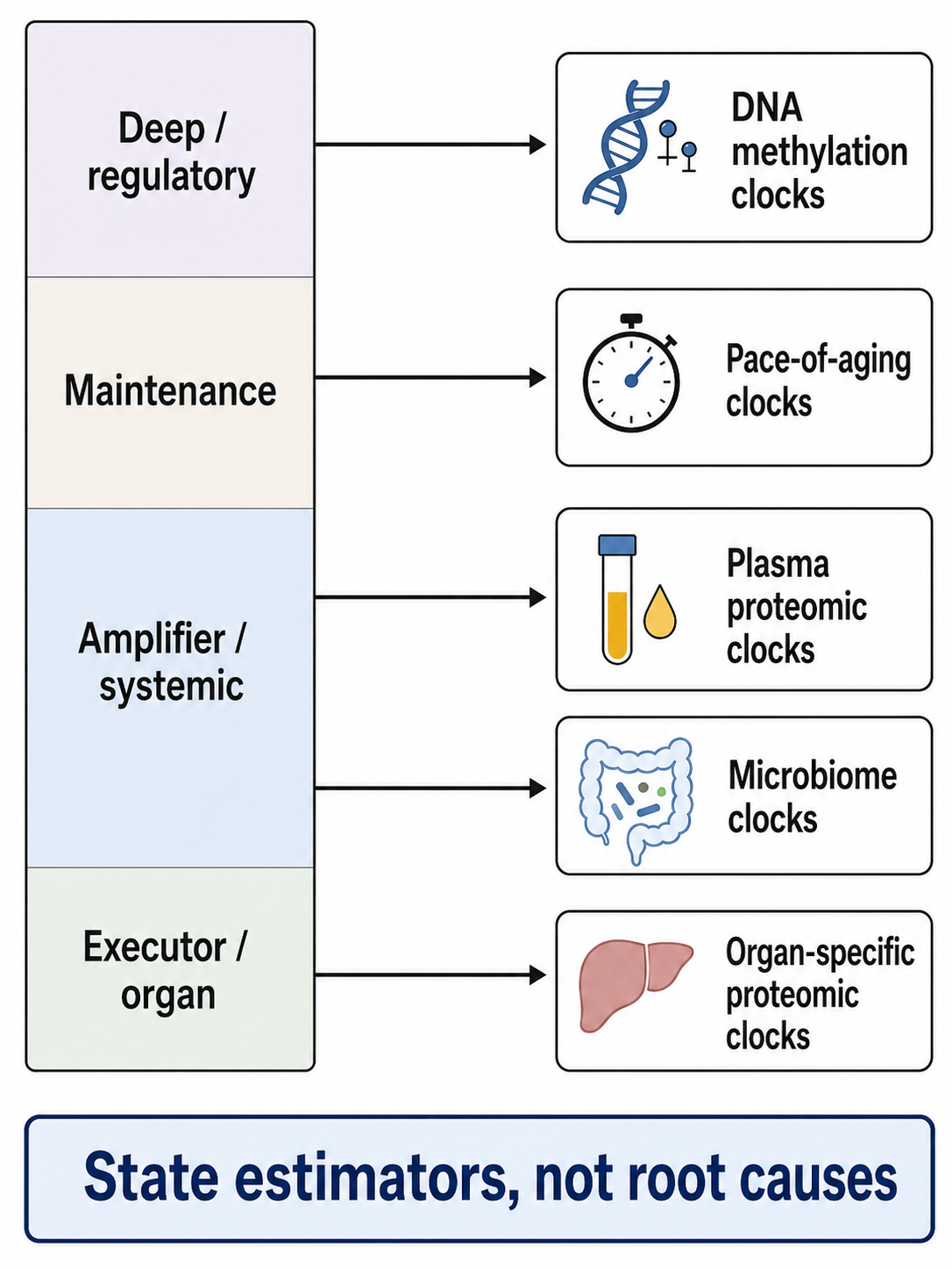
Figure 8. What clocks see. Aging clocks sample structured biological state from different layers of the system, but their predictive power does not by itself establish causal primacy. Their best use in this framework is as state estimators and falsification tools.
This point is especially important because the strongest current clocks often arise from the epigenetic layer. Since methylation signatures are conserved across tissues and mammals [18–20], strongly associated with lifespan and healthspan [109], and often move under rejuvenation-like interventions discussed earlier, it is reasonable to infer that epigenetic regulation lies unusually close to a central causal layer. Indeed, that inference is part of the larger argument of this paper. But even if epigenetic information loss is a deep mechanism, it still does not follow that the particular CpG collections used in any given clock are themselves the cause of aging. A measurement can sit close to mechanism without being identical to mechanism. The right conclusion is therefore not that clocks are “just correlations,” nor that clocks are “the mechanism.” The right conclusion is that some clocks may be privileged windows onto deeper biology while still remaining windows rather than the full architecture [4,18–20].
Once clocks are put in that epistemic place, their scientific value becomes clearer rather than smaller. They are best treated as state estimators and falsification tools. If the theory developed in this paper is correct, then interventions that restore cell-state fidelity should move epigenetic clocks more readily than they reverse deeply entrenched structural lesions. Interventions acting on rate-control pathways should affect pace measures such as DunedinPACE [110]. Interventions targeting systemic milieu should register in plasma and organ-specific proteomic models [103,114,120,121]. Organ-specific diseases should appear as selective acceleration in the most burdened systems before full organismal collapse is clinically obvious [120,121]. Microbiome manipulation should move microbial-age measurements and perhaps some systemic biomarkers without thereby proving that all upstream layers have been reset [72,73]. In this way, clocks do not replace causal theory; they constrain it. A proposed aging theory that cannot predict which biomarkers should move first, which should remain discordant, and which organs should show the earliest acceleration is not yet a sufficiently sharp theory.
Indeed, disagreements among clocks may be as informative as agreements. Convergence across methylation, proteomic, transcriptomic, and microbiome-based measures suggests that multiple biological layers are reporting a shared organismal age state [18–20,72,73,96,98,114,120,121]. Divergence suggests something equally important: that a perturbation may be acting disproportionately on a particular layer, tissue, or amplifier module. A therapy could improve inflammatory and proteomic age while leaving deeper genomic burden relatively untouched. A tissue-specific regenerative intervention could move an organ clock without immediately normalizing whole-body plasma age. A microbiome intervention could shift ecological age signals while only partly affecting systemic readouts. Such patterns are not nuisances to be averaged away. They are clues to causal structure. In the framework proposed here, discordance among clocks is a feature, not a defect, because it helps localize where in the hierarchy a perturbation is acting.
The existence of multiple successful clocks across different data types also carries a deeper implication. It suggests that aging is not merely measurable because each biomarker captures its own isolated phenomenon. Rather, many layers of biology appear to co-vary along a shared aging trajectory. Methylation, plasma proteins, microbiome composition, cell-state atlases, and multi-omic dynamics all recover age-related order from different vantage points [18–20,72,73,96,98,100,103,114,115,120,121]. That is strong evidence that aging has a genuine latent structure. But latent structure is not the same thing as causal essence. Many upstream processes can project onto the same age axis. A clock can therefore be accurate because it registers the integrated consequences of multiple coupled mechanisms, not because it has isolated the first cause among them.
That is why clocks are best understood as part of the empirical infrastructure of theory rather than as substitutes for theory. They tell us where an organism or organ appears to sit along an aging trajectory. They tell us how fast the trajectory may be moving. They help identify accelerated-aging states, latent disease vulnerability, and intervention response [109,110,115,120,121]. They can also reveal whether aging is broad or organ-restricted, smooth or nonlinear, coherent or discordant across layers [96,98,100,103,114]. But they do not, by themselves, answer the central causal questions posed in this paper: what lies deepest, what amplifies local failure into organism-wide decline, and what remains most reversible.
The proper conclusion, then, is twofold. First, clocks show that biological age is real in a stronger sense than mere chronology: it is a structured, conserved, measurable property of living systems across tissues, species, and omic layers [18–20,72,73,96,98,100,109,110,114,115,120,121]. Second, clocks do not by themselves tell us what aging is in causal terms [4]. In the hierarchy proposed here, they belong to the biomarker and readout layer. They are indispensable because they let us quantify the shadows cast by deeper mechanisms. But shadows are not the objects that cast them. The task of theory is to explain why those shadows take the form they do. The task of clocks is to let us measure whether the explanation is right.
| Readout | Best interpretation |
|---|---|
| DNA methylation clocks | Deep regulatory-state estimators; not proof that measured CpGs are root causes. |
| Pace-of-aging clocks | Tempo estimators; useful for tracking intervention effects. |
| Plasma proteomic clocks | Systemic aging-state estimators integrating signals from many tissues. |
| Organ-specific proteomic clocks | Executor and organ-vulnerability estimators before overt disease. |
| Microbiome clocks | Host–microbe amplifier estimators; context-dependent. |
| Single-cell atlas states | Cell identity and tissue-heterogeneity readouts. |
Table 8. Biomarkers and clocks as state estimators. Aging clocks sample structured biological state from different layers of the system, but predictive strength alone does not establish causal primacy.
The unified theory
The evidence assembled in the preceding sections supports a stronger claim than the familiar statement that aging is simply “multifactorial.” It supports a ranked causal architecture. The hallmarks are real recurring features of aging biology [1,2], but they do not all occupy the same logical level. Some are deep causal drivers, some are failures of maintenance, some are protective responses that become pathological, some are amplifier loops, some are tissue-specific executor phenotypes, and some are readouts. The concept that organizes these layers is biological fidelity. By this I mean not merely the storage of information in DNA, but the organism’s ability to preserve, interpret, repair, and faithfully enact biological information across scales: genomic integrity, chromatin state, transcriptional identity, proteome quality, organelle coordination, tissue architecture, and intercellular communication [5]. Aging, in this view, is the progressive failure of those fidelity-preserving systems.
Stated compactly, the theory advanced here is this: aging is the progressive loss of biological fidelity across coupled molecular, cellular, tissue, and systemic layers; conserved nutrient- and stress-sensing programs modulate the rate of that loss; antagonistic responses such as senescence and inflammation convert it into chronic pathology when repair and clearance fail; amplifier loops spread local failures into organism-wide state change; and tissue-specific executor modules determine the phenotype by which aging is finally expressed [6,18–23,29,31,33,34,38–41,44–46,50,55,57,64,65,68,71,79,85,96,98,100,117–121].
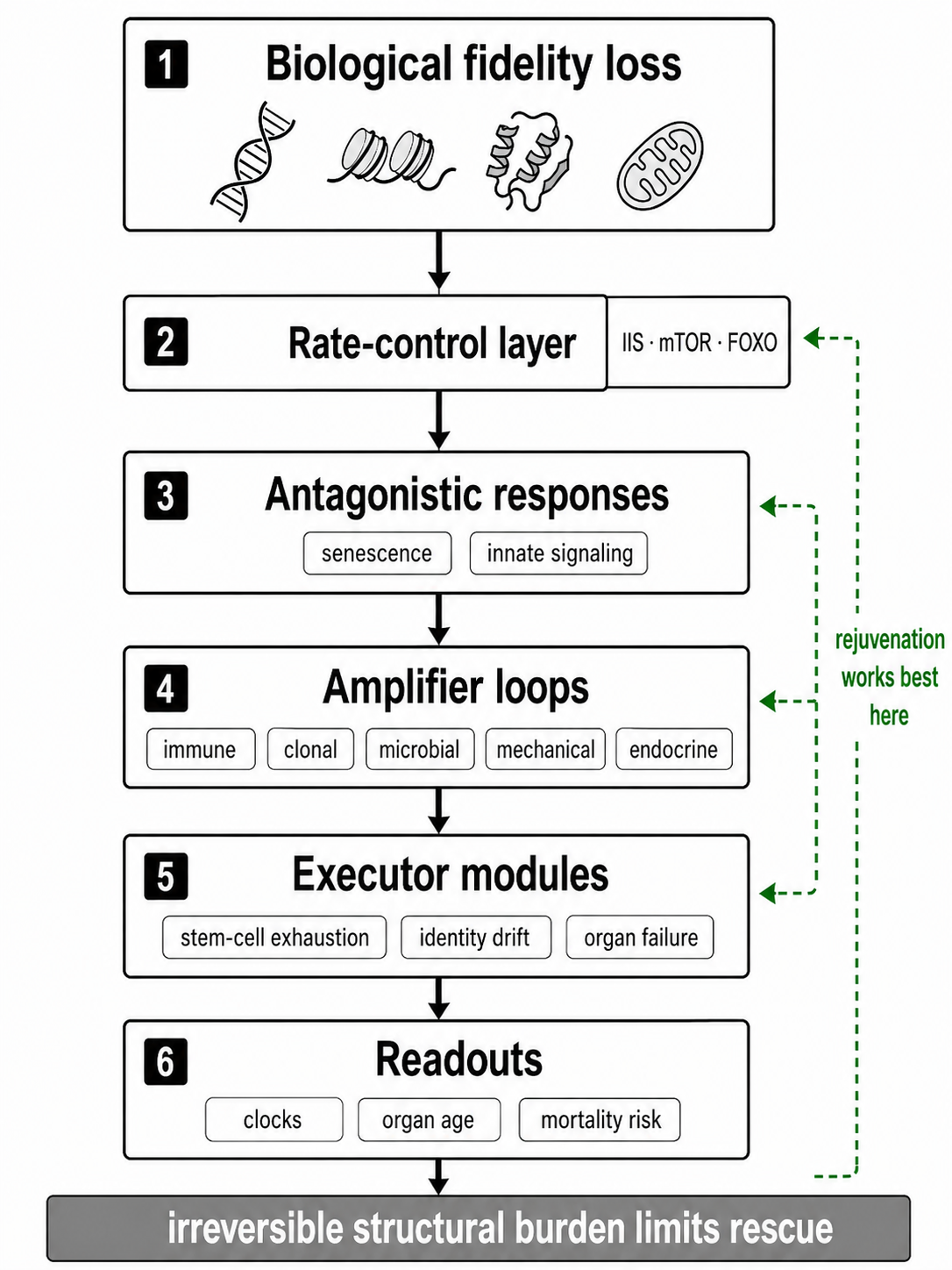
Figure 9. The unified theory. Aging is modeled as progressive loss of biological fidelity across coupled layers, paced by conserved allocation programs and converted into systemic decline by self-amplifying loops before being expressed through tissue-specific executor modules.
At the deepest level, aging begins not with any single downstream hallmark but with the gradual erosion of the templates and control systems from which living order must be continuously reconstructed. DNA damage and somatic mutation impose a hard material constraint on long-lived tissues and organisms [6,9]. Yet the most unifying evidence points beyond sequence lesions alone toward damage-driven distortion of biological information in chromatin and cell-state control. DNA methylation patterns track age across tissues and across mammals [18–20]; experimentally induced chromatin disruption can drive aging-like phenotypes [21]; and partial reprogramming can restore youthful regulatory state and function without whole-organ replacement [22,23]. Telomeres fit within this same framework as a lineage-specific boundary condition that becomes decisive in highly proliferative systems, not as a universal master clock. The key point is that living tissues must continually rebuild correct state from perturbed substrates, and aging begins when that rebuilding becomes progressively less faithful.
This fidelity loss is then propagated within cells by deterioration of the systems that convert stored biological information into working biology. Proteostasis can fail early in adult life [29], autophagy is mechanistically required for major lifespan-extending interventions [31], age-linked translational error directly corrupts the expressed proteome [117,118], and mitochondrial dysfunction, mtDNA instability, and NAD-dependent mito-nuclear uncoupling degrade energetic and signaling coherence [33,34,38,39]. This is why fidelity is a more useful term than information alone. Information may still exist in principle while its execution is corrupted at the levels of translation, protein folding, organelle quality control, and metabolic coupling. Aging is therefore not only the accumulation of damaged parts. It is the progressive reduction in the organism’s ability to generate reliable parts, states, and signals from increasingly noisy templates.
The pace of that process is not fixed. Conserved nutrient-sensing pathways sit above the maintenance layers as a rate-control layer. IIS–FOXO–mTOR–AMPK signaling does not create aging ex nihilo; rather, it regulates how aggressively organisms allocate resources toward growth, reproduction, biosynthesis, stress resistance, autophagy, and repair. The ability of daf-2 mutation, late-life rapamycin, and caloric restriction to slow aging across worms, mice, and primates shows that the tempo of fidelity loss is itself deeply regulable [40–42]. What evolution appears to shape, on this view, is not a simple death program, but a life-history allocation regime that determines how much maintenance can be sustained over time [3,40–42]. This is why neither a purely passive wear theory nor a purely programmed theory is sufficient. Material deterioration is real, but evolved physiological programs strongly influence how fast it accumulates and how long it can be buffered.
As fidelity-preserving systems falter, cells and tissues deploy antagonistic responses that are initially protective. Senescence arrests cells at risk of malignant or dysfunctional behavior; inflammatory danger signaling mobilizes defense and repair. The senescence-associated secretory phenotype allows damage signals to be broadcast beyond the original cell [101], and transient senescence can be beneficial in wound healing [102]. But these responses are not stably benign. With age, immune surveillance, clearance, and tissue context deteriorate, and what began as a local protective brake becomes a chronic source of dysfunction. Genetic and pharmacologic clearance of senescent cells improves late-life function and lifespan [44–46], while cGAS–STING activation and LINE-1 derepression show how DNA and chromatin instability can be translated into persistent inflammatory signaling [50,55]. Senescence and inflammaging are therefore not best treated as the universal root cause of aging. They are conversion mechanisms by which upstream fidelity loss becomes durable tissue pathology.
From there, aging becomes organismal through self-reinforcing amplifier loops. Clonal expansion and hematopoietic skew translate cell-autonomous variation into altered tissue composition, inflammatory tone, and disease risk [10–13,62,63]. Dysbiosis and host–microbe feedback influence immune, metabolic, and regenerative state [70–75]. ECM remodeling and tissue stiffening feed back on gene regulation, stem-cell behavior, and senescence [67–69]. Neuroendocrine and circulatory systems synchronize these local disturbances across the body, as shown by heterochronic parabiosis and hypothalamic control studies [57,64,65]. By the time these coupled loops dominate, aging is no longer reducible to isolated lesions within isolated cells. It has become a distributed systems process in which local errors are amplified, synchronized, and stabilized across tissues. This helps explain why organismal aging trajectories can show nonlinear transitions rather than smooth linear drift [100].
Tissue-specific aging emerges when this common upstream process is filtered through distinct executor modules. Organs differ in turnover rate, stem-cell dependence, immune exposure, mechanical load, barrier function, metabolic demand, and reserve capacity. As a result, the same deeper architecture yields different dominant outputs: regenerative collapse in one tissue, identity drift or mesenchymalization in another, inflammatory remodeling in another still. Single-cell atlases show that aging is both coordinated and cell-type-specific [96,98], and organ-specific aging models show that different systems can become biologically older at different apparent tempos [120,121]. The executor phenotype is often not simply cell death. It is mis-specification: stem cells that no longer regenerate correctly, immune populations that lose stable identity, cells pushed into maladaptive mesenchymal or inflammatory states, and tissues whose altered mechanics and composition lock those states in place [27,28,68,69,98,119]. Organ heterogeneity therefore does not argue against a unified theory. It is exactly what a layered architecture should produce when shared upstream failures are expressed through different tissue designs.
This framework also explains why aging is partially reversible without being indefinitely reversible. If aging were only cumulative, irreparable wear, then partial reprogramming, telomerase rescue, NAD restoration, systemic-factor manipulation, or niche correction should have limited ability to restore function. Yet such interventions can rejuvenate specific tissues, improve stem-cell performance, move biological-age measures, and in some settings extend lifespan [16,17,22–25,39,57,59,71,73,74,104,119]. These findings do not show that all damage is reversible. Rather, they show that a substantial fraction of aging resides in regulatory state, network coordination, and maintenance capacity. Rejuvenation works when it restores fidelity-preserving programs faster than irreversible structural losses—mutation burden, cell loss, fibrosis, cross-linked matrix, entrenched architectural damage—have accumulated beyond compensation. In this sense, the reversible fraction of aging lies disproportionately in the regulatory and amplifier layers, while the irreversible fraction lies disproportionately in the deepest and most structurally embodied lesions.
The main reason to prefer this theory, however, is not that it sounds comprehensive. It is that it explains the field’s hardest constraints at once. It explains cross-species tempo scaling because long-lived species appear to slow both mutational and epigenetic tempo, or better buffer their consequences [9,20,79]. It accommodates exceptional longevity because different species can reinforce different layers of the hierarchy rather than abolish aging biology altogether: bats reveal unusually slow epigenetic aging [79], naked mole rats and bowhead whales appear to strengthen extracellular, repair, and tumor-resistance strategies [77,80–82], and Hydra can suppress aging-like decline unless autophagy is experimentally compromised [83–85]. It explains accelerated-aging states because progeroid syndromes, cancer therapy, and related perturbations overload particular layers of the same architecture—nuclear architecture and DNA maintenance in some cases, chronic senescence and tissue injury in others—rather than inventing a wholly separate aging biology [86,90,91]. It explains tissue divergence because executor modules are organ-specific [96,98,120,121]. And it explains partial reversibility because regulatory and systemic layers remain more plastic than the deepest structural lesions [22–25,39,57,119].
This theory therefore rejects both flat pluralism and single-cause reductionism. Telomere attrition, proteostasis failure, mitochondrial dysfunction, senescence, inflammaging, clonal hematopoiesis, dysbiosis, stem-cell exhaustion, cell-identity drift, and organ failure are all real. But they do not occupy one plane. Telomeres are decisive in some lineages but not all. Senescence and inflammation become highly causal late in life but are not universal first movers. Proteostasis and mitochondrial decline are central maintenance failures, but they depend in part on deeper regulatory integrity. Clocks are powerful state estimators but not mechanisms in themselves [4]. A unified theory must therefore rank causal position. In the architecture proposed here, deep fidelity loss and the rate-control layer come first; the antagonistic response layer and the amplifier layers determine when local failures become organism-wide; executor modules determine phenotype; and the readout layer reveals where the organism now sits.
Seen this way, the hallmarks are not discarded; they are reclassified into a causal sequence. Deep causal layers concern genome instability, epigenetic information loss, and lineage-specific proliferative constraints. Primary maintenance layers concern proteostasis, autophagy, mitochondrial quality control, and metabolic coordination. The rate-control layer modulates how fast decline unfolds. The antagonistic response layer converts upstream disturbance into chronic pathology. Amplifier layers spread and stabilize the disturbance across the organism. Executor layers generate tissue failure. The readout layer reports the resulting state. This ranked interpretation turns the field from a catalog of co-occurring processes into a testable theory of ordered causation.
The resulting picture is neither pure wear nor pure program. It is not pure wear because conserved signaling pathways can slow aging, systemic context can reset function, and partial reprogramming can restore youthful state [22–25,40–42,57]. It is not pure program because mutational burden, DNA-repair demands, translational error, mtDNA instability, and structural tissue damage impose real material constraints [6,9,34,35,117,118]. The most accurate description is that aging is a progressive loss of biological fidelity across coupled scales, paced by evolved allocation programs and converted into systemic decline by self-amplifying loops.
That, in mature form, is the unified theory advanced in this paper. Aging begins when organisms become progressively less able to preserve and correctly execute biological information across genome, epigenome, proteome, organelle, tissue, and systemic layers. Conserved rate-control systems determine how quickly that loss accumulates. Protective responses such as senescence and inflammatory danger signaling become pathological as repair and clearance fail. Immune, clonal, microbial, mechanical, and endocrine-circulatory amplifiers propagate local failures into organism-wide state change. Tissue-specific executor modules determine how the common process becomes muscle weakness, immune failure, barrier dysfunction, fibrosis, cognitive decline, or organ disease. Clocks and multi-omic signatures measure the resulting state, but do not themselves explain it [4,96,100,109,110,120,121]. The task that follows is therefore not to ask which single hallmark is “the cause” of aging. It is to determine which interventions preserve or restore fidelity at the deepest and most leverage-rich layers, and which predicted sequences of molecular, systemic, and functional change would falsify this model if they fail to appear.
Explicit predictions and falsifiers
A theory of aging becomes scientific only when it risks being wrong. The fidelity-loss architecture proposed here does more than restate that many mechanisms participate in aging. It makes claims about temporal order, causal leverage, reversibility, comparative biology, and how biomarker systems should behave if they are readouts of layered causation rather than causes in themselves [4,109,110,114,115,120,121]. The value of this framework, therefore, lies not only in its ability to synthesize known findings, but in the specific patterns it predicts and the observations that would count against it. Because tissues differ and species solve longevity in partially different ways, no single experiment will likely decide the question. But a repeated failure of the expected causal ordering would seriously weaken the model.
1. The earliest broadly shared age-linked changes should appear in fidelity-preserving layers before overt organ decline.
If aging begins as a progressive loss of biological fidelity, then the first recurrent, cross-tissue changes should arise in the systems that preserve and enact state: chromatin organization and methylation patterning, proteostasis capacity, translational accuracy, and organelle communication. The expectation is not that every tissue will age identically, or that every early signal will be the same molecule. It is that, across tissues and organisms, the earliest common shifts should cluster in regulatory-state instability, proteome-quality stress, and mito-nuclear coordination before gross tissue dysfunction becomes manifest [20,21,29,38,39,117,118]. Longitudinal studies should therefore detect age progression first in methylation tempo, transcriptional-control stability, proteostasis stress markers, translational-error burden, and NAD-sensitive organelle coordination, while overt executor phenotypes—regenerative failure, organ fibrosis, frailty, barrier dysfunction, or clinically recognizable disease—should emerge later.
The strongest falsifier here would be a repeated demonstration that the earliest broadly shared changes are not in these fidelity-preserving systems at all, but instead in downstream phenomena such as persistent inflammaging, senescent-cell accumulation, or tissue-executor failure, with no consistent antecedent changes in chromatin state, proteostasis pressure, or organelle communication. If chronic inflammatory activation or overt organ dysfunction reliably precedes the predicted upstream changes across multiple tissues and models, then the hierarchy proposed here would be wrongly ordered.
2. Interventions that restore cell-state fidelity should reset clocks and function more readily than they reverse deep structural lesions.
If a substantial fraction of aging resides in corrupted regulatory state and in the failure of systems that faithfully re-enact biological information, then interventions that restore that state should have a distinctive profile. Partial reprogramming, NAD restoration, youthful systemic exposure, and related state-restoring manipulations should move biological-age readouts and recover function more readily than they erase high mutational burden, long-standing fibrosis, extensive cell loss, or other deeply embodied structural lesions [16,17,22–24,39,57,116,119]. The theory does not claim that “state” is infinitely plastic or that structural damage is never modifiable. It predicts an asymmetry: clocks, cell identity, and functional capacity should often shift faster and farther than irreversible lesions do. In practical terms, methylation and proteomic age should often improve before one sees major reversal of deeply embedded matrix changes, clonal architecture, or accumulated genomic damage.
This prediction is a direct challenge to a purely irreversible wear model. If interventions that restore regulatory state consistently fail to move biological-age measures or function, while interventions aimed solely at downstream structural lesions prove equally or more effective at restoring youthful state, then the central role assigned here to fidelity loss would be overstated. Likewise, if partial reprogramming, NAD restoration, or niche resetting do not preferentially improve clocks and reversible physiology relative to hard structural pathology, the theory loses one of its clearest discriminants.
3. Senescence removal should improve late-life function more than it slows the early-life rate of aging.
In this framework, senescence is not the deepest origin of aging but one of the most powerful conversion mechanisms by which upstream damage and dysregulation become chronic tissue pathology. The theory therefore predicts an age asymmetry. Senolytic or senomorphic interventions should produce their largest benefits once senescent burden has accumulated and clearance systems have deteriorated—namely, in late life or in states of accelerated damage—rather than chiefly by slowing the basal aging rate from youth onward [44–46]. That prediction follows from the view that senescence is initially protective but becomes pathological when persistence, SASP signaling, and failed clearance dominate [101,102]. The expected gains are thus strongest in late-life function, inflammation, frailty, organ-specific dysfunction, and perhaps lifespan extension from older starting points, more than in a dramatic slowing of the entire early-life aging trajectory.
A strong falsifier would be the opposite pattern: if early-life elimination or durable suppression of senescence consistently produces a major cross-tissue slowing of aging pace, far greater than late-life clearance improves late-life health, then senescence would look less like a converter layer and more like a root driver. The theory would also be weakened if transient senescence proves largely dispensable for repair and tissue integrity, because one of its core claims is that senescence begins as a beneficial antagonistic response before becoming chronically harmful.
4. Long-lived species should show slower tempo of information loss and/or stronger buffering of amplifier layers.
The theory does not predict that all long-lived species achieve longevity through the same molecular trick. It predicts something more abstract and more testable: species with exceptional longevity should preserve biological fidelity longer, lose it more slowly, or more effectively prevent local failures from being converted into organism-wide pathology. Cross-mammalian scaling of somatic mutation and methylation tempo already suggests that lifespan is linked to the pace at which some fundamental forms of informational drift accrue [9,20]. The comparative prediction, then, is that long-lived species should exhibit slower epigenetic tempo, slower accumulation of proteome corruption, better organelle quality control, or stronger damping of amplifier layers such as chronic inflammation, clonal takeover, dysbiosis, or tissue-mechanical lock-in. The exact mechanism may differ: bats appear to combine unusual longevity with distinctive methylation and immune features [78,79]; naked mole rats suggest reinforced extracellular and tumor-resistance strategies [80,81]; bowhead whales suggest enhanced repair and cancer-control programs [82]; Hydra indicates that sustained renewal and autophagy can suppress aging-like decline unless that maintenance is experimentally broken [83–85].
This is not a claim that every long-lived species must score better on every layer. It is a claim that their longevity should not be explainable without either slower tempo of fidelity loss or stronger buffering of its system-wide amplification. The theory would be seriously weakened if exceptionally long-lived species repeatedly showed ordinary or accelerated tempo of information loss, no enhancement of amplifier buffering, and no clear preservation of maintenance capacity—yet still achieved extreme longevity through mechanisms unrelated to fidelity preservation. In that case, the present framework would be too centered on the wrong variables.
5. Accelerated-aging states should map onto overloaded layers of the same hierarchy rather than create unrelated biology.
A major virtue of a causal theory is that it should explain perturbation models. If the hierarchy proposed here is correct, then accelerated-aging states should behave like natural experiments that overload particular layers of normal aging rather than invent entirely separate mechanisms. Progeroid syndromes should disproportionately load genome-maintenance and nuclear-architecture layers; therapy-related accelerated aging should disproportionately load damage, senescence persistence, and tissue-remodeling pathways; inflammatory-metabolic perturbations should load amplifier and executor layers more strongly than others [86–95]. These conditions need not mimic every aspect of ordinary aging. The prediction is modular overlap, not perfect identity. One should be able to map the dominant lesions of each perturbation onto the same hierarchy that structures ordinary aging and to see corresponding acceleration in relevant clocks, cell-state shifts, inflammatory signatures, and organ vulnerabilities.
The falsifier is straightforward. If accelerated-aging states continue to produce strong age-like clinical phenotypes but cannot be decomposed into overload of the same causal layers seen in normal aging—if, instead, they repeatedly display molecular architectures with little overlap to ordinary aging modules—then the claim that they are informative perturbations of the same biology would fail. The theory would also be weakened if progeroid and therapy-induced aging prove useful clinically but fundamentally misleading mechanistically, with no stable mapping to the ordinary aging process.
6. Organ-specific clocks should move in advance of overt disease in the systems most vulnerable to dominant amplifier loops.
Because this theory treats clocks as readouts rather than causes [4], it assigns them a specific scientific role: they should serve as falsification tools. If tissue-specific aging is the executor layer of a shared upstream process, then organ-resolved clocks should detect impending failure before disease is clinically obvious. Pace measures such as GrimAge and DunedinPACE should register altered organismal tempo [109,110], while plasma proteomic systems that infer organ-specific age should move first in the tissues most exposed to the amplifier loops dominating a given individual or condition [114,115,120,121]. Thus, one would expect immune- or vascular-system age to move early where clonal hematopoiesis and inflammaging dominate; muscle-system age to move where mitochondrial, proteostatic, and mechanical burdens dominate; and barrier- or metabolic-system age to move where dysbiosis and inflammatory-metabolic feedback are strongest. The central prediction is lead-time: organ-specific age signals should shift before overt disease in the organs that will subsequently fail.
This prediction separates the fidelity-loss theory from a purely descriptive clock framework. If organ-specific clocks only change once disease is already clinically established, or if they fail to track the systems that later become pathological, then they would look more like passive consequences than useful readouts of preclinical executor loading. The theory would be especially weakened if these clocks systematically fail to align with the dominant amplifier loops or to provide advance notice of organ-specific decline.
Taken together, these predictions define a pattern rather than a single biomarker. The theory expects: upstream fidelity-layer changes before downstream tissue collapse; greater reversibility of regulatory state than of deep structural lesions; larger late-life than early-life benefit from senescence targeting; slower tempo or stronger buffering in exceptionally long-lived species; modular mapping of accelerated-aging states onto the normal hierarchy; and organ-specific clocks that move before overt disease in the systems most at risk. None of these expectations is arbitrary. Each follows from the claim that aging is a layered process in which deep fidelity loss is paced by conserved allocation programs, converted into chronic pathology by antagonistic responses, amplified into organism-wide state change, and finally expressed through tissue-specific executors.
The most decisive experiments are therefore already implied by the theory. They include longitudinal multi-omic studies beginning before overt decline; head-to-head comparisons of state-restoring versus damage-targeting interventions; age-stratified senescence-intervention studies; cross-species measurements of informational tempo and buffering capacity; perturbation atlases that map accelerated-aging states onto ordinary aging modules; and prospective studies testing whether organ-specific clocks anticipate disease rather than merely accompany it [9,20,22–24,39,44–46,50,57,79,80,82,85,86,91,109,110,114,115,120,121]. A theory that cannot survive such tests does not deserve to be called causal. A theory that does survive them would have moved aging biology beyond mechanism lists and toward an experimentally discriminable architecture.
| Prediction | Falsifier |
|---|---|
| Early fidelity-layer changes precede organ decline | Inflammation or organ failure repeatedly appears first without upstream fidelity change. |
| State-restoring interventions move clocks and function before deep lesions | Structural lesions reverse first while regulatory state remains old. |
| Senescence targeting works best late in life | Early-life senescence suppression dominates late-life clearance effects. |
| Long-lived species buffer information loss or propagation | Long-lived species show ordinary tempo and no enhanced buffering. |
| Accelerated-aging states map to hierarchy layers | Fast-aging states repeatedly show unrelated molecular architectures. |
| Organ-specific clocks precede overt disease | Organ clocks change only after disease is clinically established. |
Table 9. Predictions and falsifiers of the fidelity-loss hierarchy. These claims make the theory experimentally testable by specifying what should be observed if the hierarchy is correct and what would count against it.
Alternative interpretations and limitations
Any theory ambitious enough to reorder the causal landscape of aging must confront the possibility that it has merely renamed old disagreements rather than resolved them. The present framework should therefore be judged not by whether it abolishes rival views, but by whether it can place their strongest insights within a more powerful causal architecture while explaining more of the comparative, mechanistic, and interventional evidence than any single alternative alone [3–5]. In that spirit, the major objections are serious and worth stating in their strongest form. Some will argue that aging is fundamentally accumulated molecular damage; others that it is chiefly programmed; others that clocks already provide sufficient descriptions; others that senescence or chronic inflammation are the true roots; and still others that animal aging is too diverse for any unified theory to be meaningful [76,84,85]. These objections do not weaken the argument by merely existing. They define the standards a useful theory must meet.
| Alternative view | Placement in this theory |
|---|---|
| Aging is accumulated damage | Damage is real, but becomes aging through loss of fidelity and coordination. |
| Aging is programmed | Rate-control programs regulate tempo, but are not a universal death script. |
| Clocks explain aging | Clocks measure structured state, but do not by themselves rank causality. |
| Senescence is the root | Senescence is a powerful converter layer, especially late in life. |
| Inflammation is the root | Inflammation is a central amplifier, not the deepest origin. |
| Species age too differently | Diversity reveals which layers can be slowed, buffered, or compressed. |
Table 10. Alternative interpretations and how the hierarchy absorbs them. The fidelity-loss theory does not deny damage, program-like regulation, clocks, senescence, inflammation, or species diversity; it assigns each to a specific causal role.
Aging as accumulated molecular damage rather than information loss.
The first alternative is that aging is, at bottom, accumulated molecular damage and that the language of “biological fidelity” or “information loss” adds little beyond metaphor. This objection has real force. Evolutionary accounts of aging already imply incomplete maintenance and repair, not perfect somatic preservation [3]. No serious theory can dispense with DNA lesions, replication errors, protein damage, organelle dysfunction, or other forms of material deterioration. In that sense, the fidelity-loss architecture is not an anti-damage theory. It accepts that lesions are unavoidable and that repair is costly, incomplete, and evolutionarily underwritten. Its claim is narrower and more specific: damage becomes organismal aging insofar as it degrades the capacity of living systems to faithfully preserve, interpret, repair, and re-enact biological state across levels [5].
That distinction matters because a pure damage-accumulation view struggles with several features of the aging literature. Mitochondrial biology is an obvious example. The decline of the classical mitochondrial free-radical theory as a complete explanation of aging, together with the rise of mitohormesis, showed that the quantity of oxidative insult alone does not determine outcome; adaptive response architecture matters decisively [36,37]. The same lesion burden can produce resilience in one context and collapse in another. That is already an argument that aging cannot be reduced to the arithmetic sum of molecular insults. Likewise, interventions that partly restore youthful regulatory state can recover substantial function without first erasing every upstream lesion, suggesting that phenotype depends not only on accumulated damage but on how faithfully cells and tissues can still execute coordinated states [24]. A damage-only theory can acknowledge such findings, but it often explains them awkwardly. The fidelity-loss framework absorbs them more naturally by treating damage as substrate and distortion of biological state as the mechanism by which that substrate becomes organism-wide decline.
This does not mean that “information loss” is automatically superior language. The objection remains useful because it warns against vagueness. If information loss is allowed to mean everything, it means nothing. The concept is only valuable when tied to measurable failures of genomic integrity, chromatin-state maintenance, proteome quality control, organelle communication, and multicellular coordination. The framework proposed here is therefore best understood not as a rejection of molecular damage, but as a claim that damage alone is too low-level and too undifferentiated to explain why aging is structured, coordinated, partially reversible, and predictably buffered in long-lived lineages [5,36,37].
Aging as primarily programmed.
A second alternative is that aging is primarily programmed. This view also captures something important. Lifespan is clearly regulable, and conserved pathways that coordinate growth, reproduction, nutrient sensing, and stress resistance do exert strong control over aging tempo. That observation has long motivated more program-like interpretations of aging biology [3]. Any adequate theory must therefore avoid the opposite mistake of describing aging as mere passive rust. Conserved control systems do exist, and they do matter.
The problem arises when rate-control is promoted into a full death program. The stronger version of the programmed view claims that aging is fundamentally a genetically specified trajectory of decline. That interpretation fits uneasily with both evolutionary logic and comparative evidence. The evolutionary literature more often supports trade-offs, antagonistic pleiotropy, and underinvestment in maintenance than a selected program whose direct function is organismal deterioration [3]. Comparative biology deepens the problem. Aging is not expressed with the same tempo, the same pathology, or even the same apparent inevitability across animals [76]. Hydra is especially difficult for a rigid programmed model: negligible senescence can be maintained in one context, yet aging-like decline appears when key maintenance systems such as autophagy are experimentally compromised [84,85]. That pattern is more consistent with conditional failure of fidelity-preserving systems than with the unfolding of a universal aging script.
The fidelity-loss architecture therefore keeps what is strongest in the programmed view while discarding what is least persuasive. It accepts that there are conserved, evolution-shaped systems that regulate how aggressively maintenance is funded, how stress responses are mounted, and how quickly fidelity is lost. But it rejects the claim that aging is simply an active program to decay. In this model, program-like regulation is real at the level of rate-control, allocation, and response, not at the level of a single master script whose aim is decline. Aging is neither pure wear nor pure program. It is deterioration occurring in a regulated organism.
Clocks as sufficient descriptions.
A third alternative is epistemic rather than mechanistic: perhaps aging clocks and multivariate state estimators already provide all the description we need, and causal theory is secondary. This position deserves respect because clocks have become extraordinarily informative. They predict morbidity, mortality, and pace with impressive accuracy, and they increasingly resolve organ-specific or system-specific aging states. Yet the central caution of the clock literature remains decisive: prediction is not mechanism [4]. A clock can summarize where an organism is in aging state-space without disclosing why it got there.
This matters because the very success of clocks can tempt the field into reification. A clock age may look like a cause when it is, in fact, a compressed readout of many underlying processes. Two individuals may arrive at similar biological-age estimates through quite different mixtures of genomic instability, inflammatory amplification, stem-cell attrition, mitochondrial dysregulation, or tissue-mechanical lock-in. Conversely, an intervention may improve a clock by restoring systemic coordination or regulatory state while leaving deeper structural lesions only partly changed. Without a causal architecture, clocks tell us that aging has occurred but not which layer should be targeted first, why one clock moves faster than another, or how to interpret dissociation between readouts [4].
The present theory does not compete with clocks. It assigns them a different role. They are state estimators, comparators across tissues and species, and tools for falsification. Their importance actually increases in a layered framework, because a good theory should predict which clocks move first, which interventions shift them, and when clock change should outpace structural repair. In that sense, the clock-centered alternative is not false so much as incomplete. It risks substituting compression for explanation.
Senescence or inflammation as the true root cause.
A fourth alternative argues that the field has already identified the true origin of aging in chronic senescence or inflammation. This view is attractive precisely because these processes sit at so many points of convergence. Chronic low-grade inflammation is now recognized as a major integrator of aging biology, connecting immune dysfunction, tissue remodeling, metabolic disease, and degenerative pathology [47–49]. Senescence is similarly compelling because senescent cells can reshape niches, alter neighboring cell states, disrupt repair, and maintain pathogenic secretory environments. From the perspective of late-life dysfunction, it is easy to see why either process might look like the fundamental cause.
The difficulty is that convergence can be mistaken for primacy. Inflammation appears central partly because many distinct upstream failures flow into it: DNA damage, mitochondrial stress, retrotransposon activation, dysbiosis, clonal hematopoiesis, tissue debris, and senescent persistence all help generate inflammaging [47–49]. The same is true of senescence. Its late-life causal power does not by itself establish that it is the earliest source of aging. Indeed, the fact that so many lesions and failures converge on senescence and inflammation is exactly why they feel so central. But convergence is not the same as origin. A central hub may be the most visible node in the network without being the deepest one.
This is why the present framework classifies these processes within the antagonistic response layer and the amplifier layers rather than as sole first causes. That classification does not minimize their importance. On the contrary, it explains why targeting them can yield large benefits even if they are not the earliest lesion. The hallmarks framework itself already groups together causes, responses, and consequences that interact strongly [1,2]. The fidelity-loss architecture sharpens that picture by asking where senescence and inflammation sit in causal sequence. Their breadth of influence is real, but their explanatory success may reflect their role as common translators of upstream damage and dysregulation into organism-wide decline rather than their status as singular roots [47–49].
Too much species diversity for any unified theory.
A fifth objection is perhaps the deepest: different species may age through too many distinct mechanisms for any unified theory to survive serious comparative scrutiny. This concern is legitimate. Animal aging is not a single mammalian story with minor ornamentation [76]. Some species show negligible senescence, some achieve exceptional longevity, and some appear to reinforce very different forms of maintenance. If unification required the same molecular trigger or the same detailed sequence in every lineage, then the project would probably fail.
But that is not the kind of unification proposed here. The theory is architectural rather than molecularly monistic. It does not claim that all long-lived species solve aging with the same genes or that all animals fail in the same tissues for the same immediate reasons. It claims that recurring causal roles can still be identified across diversity: loss of fidelity in state-preserving systems, regulatory control of pace, conversion of upstream lesions into chronic pathology, amplification into systemic state change, and execution through tissue-specific failure modules. Comparative work already suggests that the molecular signatures of longevity recur in repair, maintenance, cancer resistance, metabolic restraint, and related buffering systems, even if the precise implementations vary by lineage [77]. Hydra is again instructive, because it shows that an organism capable of negligible senescence can still exhibit aging-like decline when a key maintenance function is broken [84,85]. That is not evidence against unification. It is evidence that the correct level of unification is functional and causal rather than strictly molecular.
The diversity objection therefore forces an important limitation into the theory: any successful unified account must be abstract enough to tolerate multiple implementations. A theory that demanded one universal lesion would indeed be too rigid. The present framework tries to avoid that mistake by unifying classes of problem rather than insisting on one molecular origin for all animal aging [76,77,84,85].
Premature-aging syndromes and perturbation models are informative but incomplete.
A related issue concerns accelerated-aging states. Progeroid syndromes, therapy-induced aging, obesity-related acceleration, and chronic inflammatory disorders are immensely useful because they reveal what happens when particular layers are overloaded. Yet the literature on premature-aging diseases also repeatedly warns against overreading them [90]. These states mimic parts of normal aging, not necessarily the whole. That caution applies to the current theory as well. The framework gains support when such conditions can be mapped onto its layers, but it would be an error to assume that any single progeroid or accelerated-aging model reveals the entire architecture of ordinary aging. Their value is modular, not total. They illuminate parts of the hierarchy and help distinguish drivers from passengers, but they do not abolish the need for longitudinal study of normal aging itself [90].
Conceptual limitations of the fidelity-loss model.
The most obvious limitation of the present framework is conceptual breadth. “Biological fidelity” is a useful term only insofar as it names a measurable family of failures rather than a rhetorical catch-all [4,5]. If treated as a single hidden essence, it would become scientifically weak. The term should therefore not be understood as a claim that aging reduces to one scalar quantity of “information” in an abstract or purely Shannon-like sense. What is meant here is more concrete: the declining ability of organisms to preserve accurate genome maintenance, chromatin state, proteome composition, organelle communication, and intercellular coordination over time [5]. Even so, integrating these into a disciplined empirical program remains difficult. The theory still requires operationalization—specific assays, comparative metrics, and longitudinal thresholds—if it is to avoid vagueness.
A second limitation is that the causal hierarchy is clearest early in aging and less clean late in aging. Once decline is established, feedback loops multiply. Inflammation can worsen genomic instability; senescence can distort tissue mechanics; clonal evolution can reshape systemic milieu; mechanical drift can stabilize aged transcriptional states; organ failure can feed back on the entire organism [1,2,47–49]. For that reason, the categories of root driver, amplifier, and executor should be understood as dominant causal roles, not as permanently isolated compartments. The model is hierarchical, but not linear in the simple sense. That complexity is biologically realistic, yet it also makes causal ranking harder to prove experimentally.
A third limitation is evidentiary asymmetry. Human aging research is rich in biomarkers, longitudinal association, and heterogeneous clinical phenotypes, but relatively poor in direct mechanistic intervention. Experimental causal evidence remains strongest in short-lived models. Comparative biology is increasingly informative, yet still taxonomically sparse and heavily biased toward a few well-studied lineages [76,77]. Reprogramming and rejuvenation data are promising, but their scope, durability, and safety boundaries remain incompletely defined [24]. Premature-aging syndromes are illuminating but partial analogues [90]. In that sense, the present theory is necessarily provisional. It is an attempt to organize existing evidence into the most testable causal architecture currently available, not a claim that every layer has already been decisively ordered.
Why this framework remains preferable despite those limitations.
These objections and limitations do not make the fidelity-loss model expendable. They show what it must still earn. Among the competing views, however, it has an important advantage: it incorporates what each rival interpretation gets right without mistaking any one of those truths for the whole of aging. Damage is real, but not self-explanatory. Program-like regulation is real, but not equivalent to a universal death program. Clocks are powerful, but they remain readouts unless embedded in mechanism. Senescence and inflammation are causally potent, but much of their potency derives from their role as convergence and amplifier layers. Species diversity is genuine, but it need not abolish deeper common architecture [3–5,36,37,47–49,76,77,84,85,90].
The strongest defense of the theory is therefore not rhetorical inclusiveness but explanatory compression. It explains why aging is conserved yet variable, why it is measurable yet not reducible to measurement, why some of it is reversible while some is stubbornly embodied, why long-lived species reveal buffering rather than magical exemption, and why late-life amplifier nodes can be therapeutically powerful without being the earliest causes. The theory may still prove incomplete, or partly misordered, or overly broad in some formulations. But a good theory does not win by eliminating all alternatives. It wins by showing where each alternative belongs. That is the claim here: the fidelity-loss architecture is not valuable because it denies damage, program, clocks, senescence, inflammation, or species diversity. It is valuable because it provides the most coherent place to put all of them at once.
Conclusion
The hallmarks framework gave aging biology a shared language, and it remains indispensable as a map of recurrent processes [1,2]. But the evidence surveyed across this paper argues that a map is not yet a causal architecture. The central problem has never been whether aging involves genomic instability, epigenetic drift, proteostatic failure, mitochondrial dysfunction, inflammation, senescence, stem-cell decline, and organ-specific disease. It plainly does. The harder question is how these processes are ordered, which of them are closest to origin, which chiefly regulate rate, which mainly amplify, and which largely express the final breakdown of tissues and systems. The conclusion of this paper is that aging is best understood not as a flat list of lesions, but as a progressive loss of biological fidelity across multiple coupled layers of living organization [1,2,5].
By biological fidelity, I mean the capacity of an organism to preserve and faithfully re-enact its functional state across time: to maintain genomic integrity, stabilize epigenetic and chromatin information, protect proteome quality, coordinate organelle function, and sustain reliable communication among cells, tissues, and organs [5,9,20,23]. Aging begins when that fidelity is no longer maintained with sufficient precision. This does not mean that aging is immaterial or detached from molecular damage. On the contrary, damage is real and unavoidable. But damage becomes aging insofar as it corrupts the systems that preserve biological state and transmit that state forward. In that sense, aging is neither mere wear in the crude mechanical sense nor a single runaway pathway. It is the gradual failure of fidelity-preserving systems in an organism that must continuously repair, interpret, and coordinate itself under finite energetic and evolutionary constraints [5,9,20].
This framework also clarifies why aging is neither pure wear nor pure program. If aging were only passive deterioration, its tempo should be far less regulable than it is. Yet conserved nutrient- and energy-sensing pathways show that lifespan and healthspan can be shifted substantially by altering how organisms allocate resources between growth, reproduction, maintenance, and stress resistance [40,44]. If aging were instead a rigid genetic program of decline, it would be difficult to explain why so much of aging appears as stochastic accumulation, imperfect repair, clonal divergence, and context-dependent failure. The evidence fits better with a hybrid view: conserved regulatory programs determine how aggressively maintenance is funded and how rapidly fidelity is lost, but they do not constitute a dedicated script whose purpose is organismal decay [5,40,44]. Aging is regulated, but it is not simply programmed.
The causal importance of this distinction becomes clearer once protective and adaptive responses are placed in their proper role. Senescence, inflammatory activation, metabolic rewiring, tissue remodeling, and related responses often begin as local defenses against damage, instability, or malignant transformation. But as surveillance, clearance, and repair become less reliable, these initially protective responses persist, spread, and feed back on their own causes. Chronic low-grade inflammation is therefore not just another hallmark among equals; it is one of the main bridges by which many local failures become a common systemic syndrome [50]. Likewise, systemic milieu and interorgan signaling help convert distributed cellular errors into whole-organism state change, which is why young systemic environments can partially restore aged function in some settings [57]. Aging, in other words, becomes organismal not only because cells incur damage, but because the body increasingly fails to resolve, contain, and reintegrate local failures.
This model explains why aging is at once conserved and variable. It is conserved because all animals must solve the same broad problem: how to preserve biological fidelity in the face of entropy, damage exposure, finite repair resources, and life-history trade-offs. It is variable because the architecture of tissues, niches, repair capacity, metabolic strategy, and regenerative organization differs across species and organs. Single-cell atlases and systems-level human studies show that aging is coordinated across the organism but expressed unevenly across cell types and tissues [96,100]. Organ-specific proteomic clocks reinforce the same point from another angle: aging is not uniform even within one body, and different organs reveal different dominant execution pathways at different times [120,121]. Tissue heterogeneity, then, is not a problem for a unified theory. It is exactly what one would expect if a shared upstream logic were being executed through different biological architectures.
The same framework helps explain exceptional longevity and negligible senescence. Long-lived mammals and negligibly senescent animals do not refute general aging theory; they illuminate its boundaries and adjustable layers. Species that age slowly appear not to escape biology altogether, but to preserve repair, fidelity, and buffering more successfully, or to prevent local failures from propagating as efficiently into systemic decline [79,85]. This is why comparative biology is so informative. Exceptional species reveal which layers of the hierarchy are most suppressible, which are most tightly coupled to lifespan, and which forms of maintenance can be evolutionarily stabilized. A good theory of aging should not be embarrassed by Hydra or unusually long-lived mammals. It should be sharpened by them.
Perhaps the strongest support for the fidelity-loss framework comes from partial reversibility. If aging were nothing but irreversible accumulation of structural lesions, rejuvenation should be rare, weak, or mostly illusory. Yet partial reprogramming can restore youthful phenotypes and aspects of epigenetic state without requiring wholesale replacement of tissues, and systemic manipulations can improve regenerative function in aged contexts [23,57,119]. These observations do not imply that all damage is reversible. They imply something more specific and more important: a substantial fraction of aging lies in regulatory state, coordination, and systemic amplification, not only in terminal structural ruin [23,24,57]. This is also why clocks are so useful and why they must still be interpreted carefully. DNA methylation, plasma proteomic, and pace-of-aging measures work because aging leaves structured signatures in system state [20,110,120,121]. But their very power as summaries means they are better understood as estimators of where the organism sits in a multiscale aging process than as direct explanations of that process [4].
Taken together, the most parsimonious conclusion is this: aging is a progressive loss of biological fidelity across genome, epigenome, proteome, organelle-control systems, and multicellular signaling networks; conserved allocation and nutrient-sensing programs regulate how quickly that fidelity is lost; protective responses become chronic as maintenance and surveillance fail; self-amplifying loops then propagate local breakdown into systemic decline; and tissue-specific dysfunction emerges as the final executor layer of a common underlying process [1,2,5,9,20,40,50,57,96,100,120,121]. This view explains why aging is conserved yet diverse, why it can be slowed, why parts of it can be reversed, and why it becomes measurable before it becomes catastrophic.
If this theory is broadly correct, then the future of geroscience depends less on expanding mechanism lists than on resolving causal sequence. The decisive questions are no longer simply which hallmarks exist, but which layers fail earliest, which layers most strongly determine pace, which amplifier loops mark the transition from local damage to organismal aging, and which interventions restore fidelity before irreversible structural loss accumulates. Aging research, on this view, should aim not merely to describe decline, but to identify where fidelity is first lost, where amplification becomes self-sustaining, and where repair of state can still outrun hardening damage. That is the practical implication of a unified theory: it turns aging from a catalog of correlated deterioration into a testable hierarchy of causes, conversions, amplifiers, and executors.
The final claim of this paper is therefore simple, but it is meant to be scientifically demanding. Aging is neither a random heap of unrelated failures nor a single prewritten program. It is the progressive loss of biological fidelity in living systems, modulated by conserved life-history control and transformed into organismal decline by self-reinforcing amplifier layers. That formulation is intended not as a metaphor, but as a causal statement. Its value will depend on whether it continues to organize evidence better than rival accounts—and, most importantly, whether it generates predictions that survive contact with experiment.
References
1. López-Otín et al. The Hallmarks of Aging. Cell. 2013. PMC3836174.
2. López-Otín et al. Hallmarks of aging: an expanding universe. Cell. 2023. PMID: 36599349.
3. Review revisiting classic evolutionary theories of aging and connecting them to newer mechanistic ideas. PMC11522123.
4. Critical review arguing that aging clocks are powerful measurements but not automatically mechanistic explanations. PMC11671503.
5. Review framing aging as a loss of biological information. PMID: 38102202.
6. Review establishing genome instability as a major aging axis and linking DNA damage accumulation to functional decline across tissues. PMID: 23398157.
7. Review focusing on DNA repair capacity in aging mammalian somatic cells. PMID: 37832947.
8. Study showing that normal human tissues accumulate age-related somatic mutations in vivo. PMID: 36213345.
9. Nature paper showing that somatic mutation rates scale with lifespan across mammals. PMID: 35418684.
10. Review arguing that clonal expansion in normal tissues is a core feature of aging, not just a cancer prelude. PMID: 38623936.
11. Landmark paper linking age-related clonal hematopoiesis to adverse outcomes in humans. PMID: 25426837.
12. Study showing that TET2-mutant clonal hematopoiesis accelerates atherosclerosis in mice. PMID: 28104796.
13. Study showing that inflammatory environments can favor Tet2-mutant clonal expansion. PMID: 29195897.
14. Classic paper showing telomere shortening during serial aging of human fibroblasts. PMID: 2342578.
15. Follow-up work linking telomere length to replicative capacity. PMC50288.
16. Mouse study showing telomerase reactivation can reverse tissue degeneration in telomerase-deficient aged animals. PMC3057569.
17. Paper showing telomerase gene therapy in adult and old mice delayed aging and increased longevity. PMID: 22585399.
18. Landmark methylation-clock paper showing that DNA methylation patterns track age across many human tissues and cell types. PMID: 24138928.
19. Mammalian clock paper showing that age-linked methylation structure is conserved across mammals. PMID: 37563227.
20. Work showing that DNA methylation rates scale with maximum lifespan across mammals. PMC10798888.
21. Cell paper showing that induced DNA breaks can drive aging-like phenotypes by eroding epigenetic information. PMID: 36638792.
22. Study reporting that partial reprogramming can recover youthful epigenetic information and restore vision in a mammalian system. PMID: 33268865.
23. Mouse study reporting that gene-therapy-mediated partial reprogramming extended lifespan in old mice. PMID: 38381405.
24. Review summarizing what reprogramming-induced rejuvenation does and does not prove mechanistically. Nature Communications. 2024. Article s41467-024-46020-5.
25. Review framing aging and rejuvenation partly as changes in cellular plasticity and identity control. PMC11535162.
26. Atlas-based work arguing that aging includes loss of immune cell identity. PMID: 39143696.
27. Paper linking age-related muscle stem-cell decline to loss of heterochromatin caused by SAM depletion and showing metabolic restoration can rejuvenate the cells. PMID: 38243132.
28. Study showing that 3D chromatin reorganization helps drive B-cell aging. PMC11178499.
29. Paper showing that collapse of proteostasis is an early molecular event in C. elegans aging. PMID: 19706382.
30. Review treating aging itself as a proteostasis-collapse problem. PMID: 21441594.
31. Study showing that autophagy is required for lifespan extension by dietary restriction in C. elegans. PMC2242811.
32. Review focusing on why autophagy declines with age and what that means for causal aging pathways. PMID: 39195254.
33. Review synthesizing mitochondrial dysfunction as a major aging mechanism with consequences for energy, signaling, and cell fate. PMID: 37196864.
34. Mutator-mouse study showing that mtDNA deletions and clonal mutations can drive premature-aging phenotypes. PMID: 18311139.
35. Work linking mtDNA mutation burden to sarcopenia. PMID: 20628647.
36. Critical review of the mitochondrial free-radical theory of aging. PMID: 20021368.
37. Review developing the mitohormesis concept. PMID: 30072093.
38. Paper arguing that declining NAD+ creates a pseudohypoxic state that disrupts nuclear–mitochondrial communication during aging. PMID: 24360282.
39. Study showing that NAD+ repletion improved mitochondrial and stem-cell function and extended lifespan in mice. PMID: 27127236.
40. Classic daf-2 paper showing that a single mutation can double lifespan in C. elegans. PMID: 8247153.
41. Landmark mouse study showing that rapamycin extends lifespan even when started late in life. PMID: 19587680.
42. Rhesus-monkey study showing that caloric restriction improves health and survival. Nature Communications. 2017. Article ncomms14063.
43. Classic Hayflick paper defining the limited proliferative lifespan of human diploid cells. PMID: 14315085.
44. Transgenic mouse study showing that clearing p16-positive senescent cells delays multiple aging-associated disorders. PMID: 22048312.
45. Follow-up work showing that naturally occurring p16-positive cells shorten healthy lifespan. PMID: 26840489.
46. Paper reporting that senolytics improved physical function and increased lifespan in old age. PMID: 29988130.
47. Review arguing that chronic inflammation should be treated as a core aging process. PMID: 37329949.
48. Inflammaging review framing low-grade chronic inflammation as a systemic amplifier that connects many hallmarks. PMC6146930.
49. Review integrating immunosenescence with inflammaging. PMC5767595.
50. Study showing that cGAS–STING signaling can drive aging-related inflammation and neurodegeneration in mice. Nature. 2023. Article s41586-023-06373-1.
51. Review treating the NLRP3 inflammasome as a central inflammatory execution node in aging and age-related disease. PMID: 38317229.
52. Review on thymic involution and immunosenescence. PMID: 38735722.
53. Paper showing that LINE-1 activation can drive type-I interferon responses in senescent cells. PMID: 30728521.
54. Study showing that SIRT6 represses LINE-1 retrotransposons. PMC4185372.
55. Paper linking LINE-1 derepression in aged and SIRT6-deficient mice to cGAS-dependent inflammation and rescue with nucleoside reverse-transcriptase inhibitors. PMC6449196.
56. Study showing that senescent cells can express PD-L1 and suppress immune clearance. PMC9583718.
57. Classic heterochronic-parabiosis paper showing that exposure to a young systemic environment can rejuvenate aged progenitor cells. PMID: 15716955.
58. Foundational review on aging, stem cells, and tissue regeneration. PMID: 15725724.
59. Study showing that rejuvenation of aged muscle stem cells can restore muscle strength. PMC3949152.
60. Paper examining molecular aging and rejuvenation in human muscle stem cells. PMC2875071.
61. Study showing that a mitochondrial unfolded-protein-response checkpoint helps regulate hematopoietic stem-cell aging. PMID: 25792330.
62. Work reporting that depleting myeloid-biased hematopoietic stem cells can rejuvenate the aged immune system. PMC11870232.
63. Review linking inflammation and the stem-cell niche to hematopoietic aging. PMC8391820.
64. Paper arguing that hypothalamic NF-κB signaling helps program systemic aging. PMC3756938.
65. Study reporting that hypothalamic stem cells partly control the speed of aging through exosomal microRNAs. PMID: 28746310.
66. Paper linking hypothalamic Menin to systemic aging and cognition. PMC10019680.
67. Review arguing that extracellular-matrix dynamics are an underappreciated hallmark of aging. PMC10187690.
68. Study showing that age-related matrix stiffening can epigenetically regulate α-Klotho in cartilage aging. PMID: 36627269.
69. Paper showing that matrix stiffness can drive muscle stem-cell dysfunction through TRAIL signaling. PMC11889993.
70. Review arguing that age-related microbial dysbiosis is mechanistically relevant to aging. PMC10909992.
71. Study showing that fecal microbiota transfer from young mice can rejuvenate aged hematopoietic stem cells. PMID: 36638348.
72. Work linking microbiome features to epigenetic age acceleration and physical fitness. PMC11019127.
73. Paper building an aging clock from large-scale gut microbiome data. PMC10790003.
74. Study reporting that rejuvenating fecal microbiota transfer enhanced nerve repair in aged animals. PMID: 38599367.
75. Review on the reciprocal relationship between cellular senescence and gut dysbiosis. PMID: 39148278.
76. Comparative-biology review framing aging mechanisms in the context of species diversity. PMID: 19223603.
77. Review extracting molecular signatures of longevity from cross-species comparisons. PMID: 28800931.
78. Review arguing that bats are unusually informative models for longevity. PMID: 39365995.
79. Paper showing that DNA methylation predicts age in bats and provides insight into exceptional bat longevity. PMC7955057.
80. Review treating the naked mole-rat as a model of healthy aging. PMID: 36318672.
81. Paper linking naked-mole-rat cancer resistance to high-molecular-mass hyaluronan. PMID: 23783513.
82. Bowhead-whale genome paper identifying candidate longevity adaptations. PMID: 25565328.
83. Review explaining why Hydra is a powerful model for studying aging and non-aging. PMC4463768.
84. Work supporting negligible senescence in Hydra. PMID: 26644561.
85. Paper showing that defective autophagy in Hydra epithelial stem cells can drive aging in a normally non-senescent context. PMC6983715.
86. Review on Hutchinson–Gilford progeria syndrome and how nuclear-envelope defects generate aging-like phenotypes. PMID: 28934587.
87. Review tying HGPS to LMNA mutation biology. PMC5195863.
88. Review on Werner syndrome covering clinical features and molecular pathogenesis. PMID: 26993153.
89. Paper explicitly using Werner syndrome to illuminate mechanisms and therapies relevant to aging more broadly. PMID: 30666569.
90. Review asking what conclusions can and cannot be drawn from genetic premature-aging diseases. PMC10933081.
91. Review arguing that cancer survivors often show accelerated-aging biology. PMC10970400.
92. Work emphasizing persistent accumulation of therapy-induced senescent cells. Nature. 2025. Article s41368-025-00380-w.
93. Paper linking obesity to diseases associated with the hallmarks of aging. The Lancet Healthy Longevity. 2024. Article PIIS2666-7568(24)00087-4.
94. Review arguing that obesity and aging share core biological hallmarks ('adipaging'). PMID: 26926488.
95. Review treating HIV as a clinically relevant model of accelerated biological aging. PMID: 40231671.
96. Tabula Muris Senis: landmark single-cell atlas showing that aging is cell-type-specific, tissue-specific, and coordinated across the organism. PMC8240505.
97. Mouse aging cell-atlas analysis extracting global and cell-type-specific patterns. PMC8046488.
98. Human Cell Aging Transcriptome Atlas extending atlas logic toward human aging. PMID: 41068321.
99. Study decoding aging-dependent regenerative decline across tissues. PMID: 37898124.
100. Paper on nonlinear dynamics of multi-omics during human aging. PMC11564093.
101. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biology. 2008. PMID: 19053174.
102. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental Cell. 2014. PMID: 25499914.
103. Lehallier B, Gate D, Schaum N, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nature Medicine. 2019. PMID: 31806903.
104. Villeda SA, Plambeck KE, Middeldorp J, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nature Medicine. 2014. PMID: 24793238.
105. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001. PMID: 11292874.
106. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Current Biology. 2004. PMID: 15186745.
107. Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004. PMID: 15175753.
108. Willcox BJ, Donlon TA, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences USA. 2008. PMID: 18765803.
109. Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019. PMID: 30669119.
110. Belsky DW, Caspi A, Corcoran DL, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife. 2022. PMID: 35029144.
111. Valenzano DR, Benayoun BA, Singh PP, et al. The African Turquoise Killifish Genome Provides Insights into Evolution and Genetic Architecture of Lifespan. Cell. 2015. PMID: 26638078.
112. Shah PP, Donahue G, Otte GL, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes & Development. 2013. PMID: 23934658.
113. Bedrosian TA, Houtman J, Eguiguren JS, et al. Lamin B1 decline underlies age-related loss of adult hippocampal neurogenesis. EMBO Journal. 2021. PMID: 33300615.
114. Oh HSH, et al. Organ aging signatures in the plasma proteome track aging and disease. Nature. 2023. Article s41586-023-06802-1.
115. Argentieri MA, Xiao S, Bennett D, et al. Proteomic aging clock predicts mortality and risk of common age-related diseases in diverse populations. Nature Medicine. 2024. PMID: 39117878.
116. Yang N, Occean JR, et al. A hyper-quiescent chromatin state formed during aging is reversed by regeneration. Molecular Cell. 2023. PMID: 37116496.
117. Shcherbakov D, Teo Y, Boukari H, et al. Premature aging in mice with error-prone protein synthesis. Science Advances. 2022. PMID: 35235349.
118. Böttger EC, et al. Translational error in mice increases with ageing in an organ-dependent manner. Nature Communications. 2025. PMID: 40021653.
119. Lu JY, Tu WB, Li R, et al. Prevalent mesenchymal drift in aging and disease is reversed by partial reprogramming. Cell. 2025. PMID: 40816266.
120. Wang Y, Xiao S, Liu B, et al. Organ-specific proteomic aging clocks predict disease and longevity across diverse populations. Nature Aging. 2025. PMID: 41299092.
121. Goeminne LJE, Vladimirova A, Eames A, Tyshkovskiy A, Argentieri MA, Ying K, Moqri M, Gladyshev VN. Plasma protein-based organ-specific aging and mortality models unveil diseases as accelerated aging of organismal systems. Cell Metabolism. 2025. PMID: 39488213.
How to Cite this Article
APA:
Negron, H. (2026, May 7). A unified multiscale theory of animal aging. Jivaro Research. https://jivaro.net/content/research/r7k4m2a
Chicago:
Negron, Harry. “A Unified Multiscale Theory of Animal Aging.” Jivaro Research, May 7, 2026. https://jivaro.net/content/research/r7k4m2a
MLA:
Negron, Harry. “A Unified Multiscale Theory of Animal Aging.” Jivaro Research, 7 May 2026, https://jivaro.net/content/research/r7k4m2a.